ОСТЕОАРТРИТ: ФУНДАМЕНТАЛЬНІ ТА ПРИКЛАДНІ АСПЕКТИ ЕТІОПАТОГЕНЕЗУ ЗАХВОРЮВАННЯ. НІЧОГО НЕ СТОЇТЬ НА МІСЦІ
Резюме. Резюме. Стаття присвячена сучасній парадигмі остеоартриту (ОА), що базується на отриманих доказах запальної теорії захворювання. ОА віддавна вважається «хворобою зношування», що призводить до втрати суглобового хряща. ОА розглядався як наслідок процесу механічного перевантаження суглоба і уразливості хрящового матриксу. Прогрес молекулярної біології 90-х років ХХ ст. дозволив змінити цю парадигму. Відкриття того факту, що численні розчинні медіатори, такі як простагландини і цитокіни, здатні підвищувати продукцію матричних металопротеїназ хондроцитами, призвело до створення «запальної» теорії ОА. Утім знадобилося ще одне десятиліття, щоб синовіт був визнаний найважливішим проявом ОА. Сьогодні патогенез ОА бачиться як анормальне ремоделювання тканин суглоба (кісткової, хрящової, синовіальної, сполучнотканинної), що ініціюється і визначається прозапальними медіаторами з розвитком метаболічної, а пізніше й функціональної дисфункції всіх суглобових структур. Джерелом прозапальних медіаторів є субхондральна кістка і синовіальна оболонка. Відкриття патогенетичних механізмів виникнення та прогресування ОА підкреслило неоднорідність цього захворювання. Було виділено етіопатогенетичні та клінічні фенотипи захворювання, покликані зіграти важливу роль у стратифікації пацієнтів на групи для поліпшення результатів лікування.
Остеоартрит (ОА) является наиболее распространенной формой ревматических заболеваний суставов и одной из основных причин боли и инвалидности у пациентов среднего и пожилого возраста [4, 7]. ОА часто называют дегенеративно-дистрофическим заболеванием суставов; еще в 1911 г. T. Muller предложил выделить группу заболеваний суставов дегенеративно-дистрофической природы как «arthrosis deformans», где ОА отводилось ведущее место. ОА долгое время называли «деформирующий остеоартроз», хотя понятно, что любое хроническое заболевание суставов в процессе развития сопровождается дефигурацией и деформаций. Положение об ОА как о дистрофически дегенеративном заболевании суставов полностью сформировалось в середине 1980-х годов, когда было определено, что хондроциты имеют низкую метаболическую активность и не способны восстановить поврежденный хрящ. Вследствие отсутствия васкуляризации хрящ также не может ответить обычной воспалительной реакцией на раздражение [8]. В последние годы четко доказано, что такой взгляд является ошибочным: ОА не просто болезнь износа (болезнь «wear and tear»), а скорее анормальное ремоделирование суставных тканей, управляемое множеством провоспалительных факторов, продуцируемых, прежде всего, субхондральной костью и синовиальной оболочкой [2, 12]. Анормальное/патологическое ремоделирование впоследствии формирует метаболическую, а позже и функциональную дисфункцию всех суставных структур [25] с развитием типичной клинической картины и однотипных патобиохимических изменений. Местная продукция провоспалительных цитокинов способна также оказывать влияние на инициацию и усугубление других возрастзависимых и метаболических заболеваний. Не зря ОА относят к заболеваниям с одним из самых высоких индексов коморбидности [5]. Недопонимание многих существенных моментов ОА проявляется, в частности, в том, что в настоящее время в странах СНГ, в том числе и в Украине, одну и ту же болезнь называют то «остеоартрозом», то «остеоартритом», то «деформирующим артрозом» [6]. ОА обычно описывается как «невоспалительное заболевание» с целью отличить его от истинно «воспалительных» заболеваний — ревматоидного артрита и серонегативного спондилоатрита. Несмотря на это, воспаление все чаще и чаще рассматривается как ведущий фактор развития основных симптомов ОА и его неуклонного прогрессирования [38]. Утренняя тугоподвижность и проходящая скованность, острые воспалительные вспышки, характеризующиеся припуханием суставов, местным повышением температуры тела, воспалительной дефигурацией, иногда покраснением суставной зоны, не являются редкостью в клинической картине болезни. Нестероидные противовоспалительные препараты (НПВП) оказываются обычно более эффективными, чем простые анальгетики или даже парацетамол [16]. Внутрисуставные инъекции глюкокортикоидов показаны не только в период острых воспалительных вспышек (реактивный синовит), но и как поддерживающая терапия [29]. Одновременно получены гистологические и радиологические подтверждения существования синовита, а также низкоуровневого воспаления, при ОА. Результаты многочисленных исследований свидетельствуют, что наличие синовита, который визуализируется при артроскопии, магнитно-резонансной терапии (МРТ) или ультразвуковом исследовании (УЗИ), может выступать суррогатным маркером тяжести течения ОА и связано с риском прогрессирования заболевания [32].
Системное повышение уровня С-реактивного белка с высокой чувствительностью отображает воспаление синовиальной оболочки у пациентов с ОА и связано с уровнем болевого синдрома [31, 40]. Отметим, что синовиальное воспаление достаточно частое и при травматическом повреждении менисков и ассоциируется с уровнем боли и дисфункции сустава [35].
Вопрос, почему синовиальная оболочка воспаляется при ОА, остается открытым. Наиболее приемлемой является гипотеза о том, что деградированные хрящевые фрагменты раздражают синовиальную оболочку, вызывая продукцию медиаторов воспаления. Синовиальные макрофаги продуцируют многочисленные провоспалительные медиаторы, в результате чего нарушается баланс репарации и деградации хряща с преобладанием последней. Два основных цитокина вовлечены в патологический процесс при ОА: интерлейкин (ИЛ)-1 и фактор некроза опухоли (ФНО)-α, которые продуцируются активированными синовиоцитами и мононуклеарами. При ОА вырабатывается недостаточно антагониста ИЛ-1, чтобы блокировать провоспалительный цитокин. Синовиальная оболочка также продуцирует повышенное количество ИЛ-6, ИЛ-10, гранулоцитарно-макрофагального колониестимулирующего фактора, а также молекулы сосудистой и межклеточной адгезии [37]. Провоспалительные медиаторы активируют хондроциты поверхностного слоя хряща, что приводит к повышенному синтезу матричных металлопротеиназ (ММП) и в конечном счете — к усилению деградации суставного хряща. Кстати, именно открытие этого факта — повышение продукции ММП под воздействием провоспалительных цитокинов, высвобождаемых синовиальной оболочкой, — положило начало «воспалительной» парадигме ОА.
Значение ММП при ОА интенсивно изучается. На сегодняшний день установлено, что ММП задействованы не только в деградации хрящевой ткани, но участвуют и в поддержании гомеостаза нормального хряща. Экспериментальные данные свидетельствуют о том, что матричная рибонуклеиновая кислота ММП-1, -3 и -13 постоянно экспрессируется в суставном хряще. Кроме того, ММП и продукты деградации аггреканов присутствуют в синовиальной жидкости здоровых людей [26, 27]. В исследовании, в котором применяли ингибиторы различных ММП, показано, что ММП-13 играет важнейшую и ведущую роль в деградации коллагена II типа [19]. Данный энзим в 5–10 раз активнее в отношении коллагена II типа, чем ММП-1 и -8 [18].
Отметим, что ММП-13 является «топически привязанным» ферментом, то есть синтезируется преимущественно в глубоких слоях хряща больных ОА, в то время как ММП-1, например, экспрессируется главным образом в поверхностных зонах [22]. Возможно, это имеет какое-то дополнительное значение при развитии заболевания? [3].
Провоспалительные цитокины также вызывают синовиальный ангиогенез и способствуют дальнейшей продукции медиаторов воспаления синовиальными клетками — так замыкается порочный круг [9] (рис. 1). Воздействие на хондроцит аномальных внеклеточных стимулов, таких как провоспалительные синовиальные цитокины, матричные изменения, паракринные и аутокринные факторы, индуцируют множество аномальных клеточных ответов, приводя к изменениям катаболизма, анаболизма, апоптозу и изменениям клеточного фенотипа, который касается дифференцирующихся клеток, гипертрофированных хондроцитов и клеток — предшественников хондроцитов.
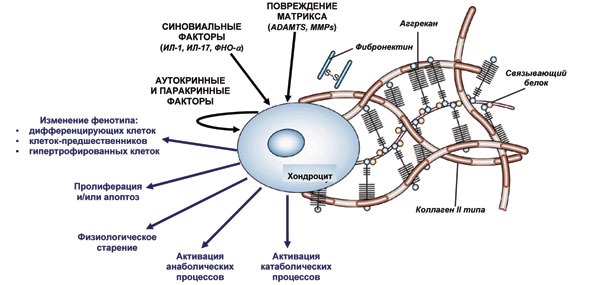
Рис. 1. Реакция хондроцитов на воздействие внеклеточных стимулов при ОА (адаптировано нами по: Aigner T. et al., 2007 [9])
Воздействие на хондроцит аномальных внеклеточных стимулов, таких как провоспалительные синовиальные цитокины, матричные изменения, паракринные и аутокринные факторы, индуцирует множество аномальных клеточных ответов, приводя к изменениям катаболизма, анаболизма, апоптозу и изменениям клеточного фенотипа
Последние данные свидетельствуют, что важным фактором, определяющим развитие и прогрессирование ОА, является врожденный иммунитет. В синовиальной жидкости у пациентов с ранними стадиями ОА установлен повышенный ответ фибробластоподобных синовиоцитов на TLR-2 (toll-like receptors) и TLR-4 лиганды, которые являются основными сигнальными рецепторами врожденной иммунной системы [30]. Повышенные уровни ИЛ-15 определяют в синовиальной жидкости при раннем ОА коленного сустава по сравнению с терминальной стадией ОА, а число CD8 синовиальной мембраны коррелирует с ММР-1 [36]. Участие toll-like receptors в патогенезе ОА схематически представлено также на рис. 2.
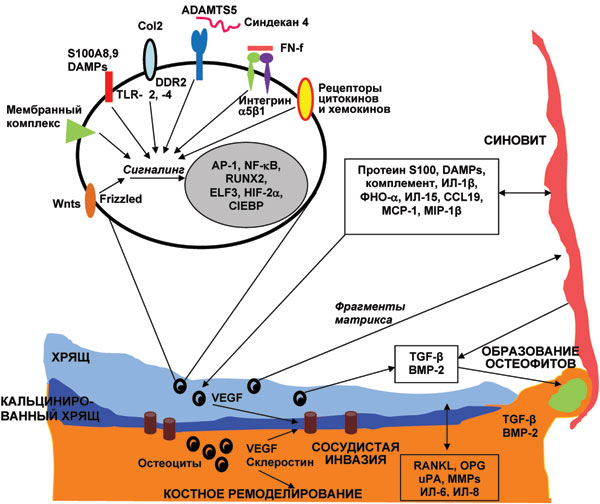
Рис. 2. Факторы суставного хряща, синовиальной оболочки и субхондральной кости, вовлеченные в остеоартритический процесс (адаптировано нами по: Loeser R.F. et al., 2012 [25]).
Белки, в том числе протеин S100 и молекулярные паттерны, ассоциированные с повреждением (DAMP), цитокины (ИЛ-1β, ФНО-α и ИЛ-15), хемокины (CCL1, моноцитарный хемотаксический протеин (MCP-1) и моноцитарный воспалительный протеин (MIP-1β)), компоненты комплемента, продуцируемые клетками синовиальной оболочки, могут стимулировать хондроциты путем активации различных рецепторов клеточной поверхности, в том числе TLR-2, -4, цитокинов и хемокинов. Другие факторы, которые активируются вследствие разрушения хрящевого матрикса, включают связывание коллагена II типа мембранным белком — дискоидин доменным рецептором (DDR2), фрагментов фибронектина (FN-f) — α5β1 интегрином, протеинов Wnt — Frizzled белками, экстрацеллюлярных факторов — синдеканом 4 (трансмембранным протеогликаном). Синдекан 4 может также быть связывать ADAMTS-5 на поверхности клетки. Различные сигнальные белки, включая активатор протеина-1 (AP-1), ядерный фактор (NF)-kВ, транскрипционный фактор (RUNX-2), Elf-3, CCAAT/усилитель связывания протеин-β (С/ЕВРβ) и гипоксия-индуцибельный фактор-2α, приводят к активации множества факторов транскрипции, которые регулируют экспрессию матричных протеиназ, индуцирующих воспаление и деградацию суставного хряща. Фрагменты разрушенного хряща, в свою очередь, могут также стимулировать развитие синовита. Продукция сосудистого эндотелиального фактора роста (VEGF) в хряще и кости стимулирует сосудистую инвазию субхондральной кости и в зоне кальцинированного хряща. VEGF, склеростин, RANKL, остеопротегерин (OPG), урокиназный активатор плазминогена (uPA), ММР, ИЛ-6 и ИЛ-8 опосредуют костное ремоделирование и потенциально способствуют разрушению суставного хряща. Трансформирующий фактор роста (TGF-β) и костный морфогенетический белок-2 (ВМР-2), продуцируемые синовиальной оболочкой, хрящевой и костной тканью, обусловливают образование остеофитов
Кроме того, активация врожденного иммунитета может быть вызвана отложением кристаллов в полости сустава [33]. Чаще всего происходит отложение кристаллов пирофосфата кальция и основного фосфата кальция. Эти кристаллы вместе с мочевой кислотой могут взаимодействовать с NALP-3 инфламмасомами, что представляет собой внутриклеточный белковый комплекс, участвующий в активации ИЛ-1 и ИЛ-18 [20, 28]. Это хорошо описано при подагрическом воспалении, однако их участие в остеоартритическом процессе вызывает дискуссию [15].
Спор о возможном только механическом повреждении суставного хряща был фактически прекращен после открытия внутриклеточного механосигналинга [23]. Оказалось, что любое ненормальное механическое напряжение — растяжение, сжатие, компрессия, напряжение сдвига, гидростатическое давление — может быть преобразовано во внутриклеточные сигналы посредством возбуждения механорецепторов, расположенных на поверхности клеток сустава. Эти сигналы могут привести к избыточной экспрессии провоспалительных медиаторов, таких как простагландины, цитокины, хемокины, в хондроцитах и клетках субхондральной кости [34, 39]. Преобразование механического сигналинга в синтез медиаторов воспаления опосредуется активацией внутриклеточных сигнальных путей, прежде всего через NF-kВ и МАРК (митоген-активируемая протеинкиназа) [10].
Таким образом, в последнее время идет активное накопление данных об инициальной и ведущей роли воспалительных медиаторов в развитии и прогрессировании ОА. Источником таких медиаторов могут быть как клетки сустава, так и другие ткани организма, прежде всего жировая ткань (особенно у пациентов с ожирением и метаболическим синдромом), которые поступают в системный кровоток и достигают сустава через субхондральную кость. Эти провоспалительные медиаторы в дальнейшем определяют ремоделирование хрящевой, костной и синовиальной тканей [12]. Правильная дефиниция ОА послужит как более точной диагностике, так и конструктивной патогенетической терапии данного заболевания. Термин «остеоартроз» останется частным понятием, отражающим конечные метаболические дегенеративно-деструктивные изменения в суставе [1].
Итак, результаты последних исследований свидетельствуют, что ОА необходимо рассматривать скорее не как дегенеративный процесс, а как анормальное ремоделирование тканей сустава (костной, хрящевой, синовиальной, соединительнотканной), определяемое провоспалительными медиаторами, источником которых могут быть суставной хрящ, синовиальная оболочка и субхондральная кость [25]. Чтобы правильно понимать патофизиологию ОА, необходимо признать, что инициация повреждения хряща и его последующее ремоделирование определяются различными самостоятельными механизмами, возможно, независимыми на ранних этапах ОА, но тесно переплетающимися в поздних его стадиях [2].
ОА является прогрессирующим заболеванием, которое характеризуется поражением всех суставных структур. Еще в 2005 г. в одном из определений ОА, предложенном OARSI, прозвучало, что «…остеоартрит — это группа патогенетически близких заболеваний, но таких, что имеют различную этиологию, однако приводят к однотипным биохимическим и клиническим последствиям…». В этом плане ОА не является уникальным заболеванием, но представляет собой гетерогенный синдром с различными клиническими фенотипами, которые постоянно развиваются, модифицируются и в конечном итоге приводят к общим клиническим проявлениям [17]. Клинические проявления заболевания зависят от формирующегося клинического фенотипа, что, в свою очередь, определяется основным патогенетическим путем поражения суставных тканей и доминирующего поражения тканей и структур сустава в определенный момент времени [1, 13, 24].
Наиболее общие факторы риска ОА включают возраст, пол, предыдущие травмы суставов, ожирение, генетическую предрасположенность и механические факторы, включая дисплазию и недостаточность связочного аппарата [14]. Однако, несмотря на многофакторность природы ОА, патологические изменения в пораженных суставах имеют общие черты, влияющие на все суставные структуры, обусловливая развитие типичной клинической картины ОА — боли, деформаций и нарушения функции.
Патологические изменения, наблюдаемые при ОА (см. рис. 2), включают деградацию суставного хряща, изменения субхондральной кости с образованием остеофитов, персистирующее, разноуровневое воспаление синовиальной оболочки, дегенерацию связок и менисков, гипертрофию суставной капсулы. Одновременно наблюдаются изменения в периартикулярных тканях: мышцах, нервах, связках, жировой ткани, которые способствуют формированию характерных симптомов ОА. Таким образом патологические изменения в суставных тканях происходят синхронно, параллельно, носят однотипный характер, что является аргументом в пользу рассмотрения ОА как болезни сустава как органа [25]. Четко продемонстрировано (см. рис. 2), как активация одних и тех же медиаторов воспаления и белковых молекул приводит к вовлечению в патологический процесс и хряща, и синовиальной оболочек, и субхондральной кости.
Однако различные этиопатогенетические факторы могут способствовать формированию определенных патогенетических и клинических фенотипов. На сегодня предложены различные фенотипические варианты течения ОА, которые затрагивают факторы риска, этиологические и патогенетические механизмы, а также клинические особенности. Так, один из предлагаемых интегрированных взглядов на патогенез и клинические проявления ОА представлен на
рис. 3. Согласно ему, выделяют первичный ОА с тремя подвидами — генетически детерминированный, эстрогензависимый и возрастассоциированный, а также вторичный ОА — посттравматический и эндокринный [42], куда можно отнести, в частности, диабет-индуцированный ОА [11].
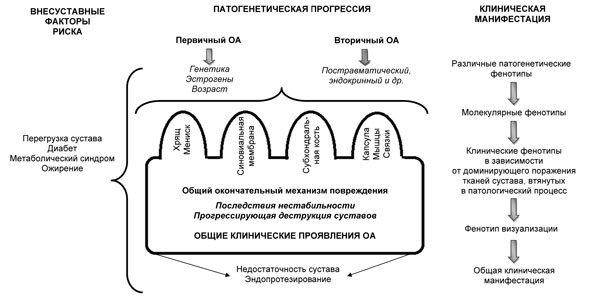
Рис. 3. Этиопатогенетический и клинический интегрированный взгляд на прогрессирование ОА (адаптировано нами по: S. Castañeda et al. (2014) [17])
ОА оказывает влияние на все суставные ткани, что приводит к различным клиническим фенотипам в зависимости от наиболее поврежденной ткани в данный момент. Когда травма субхондральной кости вследствие длительной перегрузки сустава является ведущим повреждающим фактором, то в клинической картине будут доминировать симптомы боли в костях вследствие субхондральной ишемии или отека; последний симптом отлично визуализируется при МРТ [32]. При поражении синовиальной оболочки в клинической картине будет превалировать воспалительный фенотип с частыми эпизодами реактивного синовита, воспалительным характером болевого синдрома. В других случаях изменения мягких околосуставных тканей проявляются симптомами тендинита, тендовагинита или бурсита. То есть доминирующий механизм повреждения может проявляться через различные клинические фенотипы. Позже, с течением времени и прогрессированием ОА, симптомы становятся более однородными, что описывается в литературе как «общий остеоартритический синдром» на поздних стадиях заболевания, что проявляется болью, недостаточностью сустава, нестабильностью и деформациями [17].
Различные радиологические фенотипы также коррелируют с клиническими проявлениями. Так, атрофический вариант (сужение суставной щели, остеопоротические изменения субхондральной кости) и гипертрофический (преобладание остеофитоза) ассоциируются с различными клиническими вариантами ОА. Предложенная этиопатогенетическая классификация ОА [17] обеспечивает научную базу, позволяющую понять, почему клинические фенотипы могут изменяться в течение заболевания (см. рис. 3).
Согласно другому исследованию [12], наиболее распространенными фенотипическими разновидностями ОА являются возрастассоциированный вариант, эстрогензависимый, генетически детерминированный и посттравматический. Так, возрастассоциированный вариант связан с повреждением хондроцитов и экстрацеллюлярного матрикса, уменьшением толщины и плотности субхондральной кости, саркопенией и снижением репаративной способности хряща, жесткостью сухожилий, повышением нестабильности суставов с возрастом. Наличие генов предрасположенности к ОА, низкой костной массы и формы скелета лежит в основе генетически-детерминированного варианта ОА. Кроме того, объем хряща и степень прогрессии ОА зависят от определенных генов. Общеизвестно, что снижение уровня эстрогенов вследствие менопаузы обусловливает интенсивный костный обмен в субхондральной кости, снижение костной массы, уменьшение мышечной массы и ее силы, повреждение хондроцитов и экстрацеллюлярного матрикса. Дефицит эстрогенов способствует повышению нестабильности суставов, а также увеличению массы тела, а повышенная жировая масса ассоциируется с высоким уровнем адипокинов. Кроме того, выделяют кристалл-индуцированный фенотип ОА, возникновение и прогрессирование которого могут быть связаны с нарушениями врожденного иммунитета, а также кристалл-индуцированным воспалением. Метаболический вариант [42] развивается у пациентов с ожирением, атеросклерозом, другими метаболическими расстройствами и в большинстве случаев является отображением системных нарушений метаболизма, что через провоспалительные медиаторы реализуется в суставе. Одним из вариантов метаболического фенотипа является диабет-индуцированный ОА [11]. Вследствие высокой глюкозотоксичности происходит накопление конечных продуктов гликолиза, активация оксидативного потенциала на локальном уровне, а также развитие низкоуровневого воспаления на организменном уровне [21]. Эти факторы обусловливают анормальное ремоделирование костной, суставной и синовиальной тканей, приводя к биохимическим изменениям, которые впоследствии транспонируются в клинические проявления ОА. Диабетическая нейропатия также может быть одним из повреждающих механизмов, что приводит к поражению периферической нервной системы при ОА, обусловливая слабость мышц и нестабильность сустава [41].
Главной целью попыток выделить фенотипические варианты течения ОА считают, прежде всего, индивидуализацию лечения ОА, поскольку различные патогенетически-клинические фенотипы ОА требуют различных терапевтических стратегий. Так, согласно другим исследованиям [24], было выделено еще несколько фенотипов ОА коленного сустава на основе четырех клинически значимых характеристик: рентгенографический ОА, мышечная сила, индекс массы тела, наличие и выраженность депрессии. Таким образом, было выделено пять клинических фенотипов:
- Фенотип с минимальным поражением суставов (Minimal joint disease phenotype).
- Фенотип с сильными мышцами (Strong muscle phenotype).
- Фенотип без ожирения и со слабыми мышцами (Nonobese and weak muscle phenotype).
- Фенотип с ожирением и слабыми мышцами (Obese and weak muscle phenotype).
- Депрессивный фенотип (Depressive phenotype).
Эта классификация пяти фенотипов уменьшает неоднородность ОА коленного сустава в популяции и позволяет персонифицировать немедикаментозное и медикаментозное воздействие. Общеизвестно, что именно состояние мышц, количество пораженных суставов, наличие структурных повреждений, повышенная масса тела и депрессия являются важными факторами, определяющими прогноз и эффективность лечения ОА. Представленные фенотипы могут включать различные патофизиологические и этиологические подтипы ОА. В минимальном фенотипе заболеваний суставов структурные изменения вряд ли присутствуют, симптомы обычно нерезко выражены и быстро регрессируют. Сильный мышечный фенотип может быть характерным для пациентов с посттравматическим ОА и тяжелыми рентгенологическими проявлениями, однако эти пациенты физически активны, поскольку сохраняется достаточная мышечная сила. Авторы подчеркивают, что именно при этом фенотипе чаще всего применяли артроскопические методы лечения (56% пациентов по сравнению с другими фенотипами — 22–36%; р<0,05). Выделяют также два слабомышечных фенотипа с ожирением и без ожирения. Фенотип без ожирения и слабыми мышцами чаще всего свойственен пациентам старших возрастных групп и может представлять вариант возрастзависимого фенотипа ОА. Он ассоциируется с саркопенией, остеопорозом, высокой склонностью к падениям, синдромом общей слабости и требует совершенно других подходов к лечению, чем предыдущие фенотипы. В основе формирования фенотипа с ожирением и слабыми мышцами может лежать нарушение биомеханики сустава, вызванное постоянной перегрузкой сустава вследствие повышенной массы тела. Кроме того, метаболические нарушения, связанные с ожирением, способны формировать подтип с низкоуровневым воспалением и высокой коморбидностью. Пациенты депрессивного фенотипа демонстрируют высокий уровень болевого синдрома, высокий процент нейропатического компонента болевого синдрома, нередко извращенную нервную сенситизацию, что приводит к резкому ограничению физической активности и нередко низкой приверженности лечению. Данный фенотип может представлять подтип ОА коленного сустава, при котором депрессия привела к более активному привлечению бользависимых стимулов, повышению сенсибилизации центральной нервной системы на болевые раздражители и отсутствие физической активности.
Выделение дифференциальных отличий клинических фенотипов предполагает и возможные различные клинические исходы. Наличие только одного фактора риска обусловливает незначительную прогрессию и функциональные нарушения. Множественные факторы риска (ожирение, мышечная слабость, структурные нарушения) приводят к более выраженной клинической картине и значительным функциональным ограничениям. Психосоциальные факторы также имеют значительное воздействие на клинический исход ОА коленного сустава (депрессивный фенотип). Каждый из представленных фенотипов поможет в выборе правильной тактики лечения ОА, учитывая значительную гетерогенность данного заболевания. Например, у пациентов с депрессивным фенотипом может быть эффективным сочетание образовательной программы, когнитивно-поведенческой терапии, индивидуальные занятия лечебной физкультурой, а также применение антидепрессантов. У лиц с ожирением и низкой мышечной силой, возможно, потребуется фокусирование на программах уменьшения массы тела и укрепления мышц. Одновременно для лиц без ожирения и со слабомышечным фенотипом достаточно будет только упражнений для укрепления мышц. Большинство же лиц сильного мышечного фенотипа вряд ли выиграют от лечебной физкультуры.
В одном из последних исследований, посвященных клинической гетерогенности ОА, выделение фенотипов течения ОА базируется на вариантах ОА — моноартикулярное или полиартикулярное поражение, а также факторе коморбидности, что в большинстве случаев определяет терапевтические возможности лечения [29]. Так, выделено четыре фенотипа ОА:
- Остеоартрит коленного сустава без коморбидности.
- Остеоартрит коленного сустава с коморбидностью.
- Множественное поражение суставов без коморбидности.
- Множественное поражение суставов с коморбидностью.
При этом также впервые было обозначено средний и высокий риск коморбидности при ОА. Так, к среднему риску были отнесены пациенты с ОА, имеющие диабет, артериальную гипертензию, кардиоваскулярные заболевания, почечную недостаточность, осложненные гастроинтестинальные заболевания, депрессию, физические ограничения, вызванные патологическими состояниями, в том числе ожирением. Лица пожилого возраста также были отнесены к данной подгруппе. Высокий риск коморбидности формировали пациенты с желудочно-кишечными кровотечениями в анамнезе, перенесенным инфарктом миокарда и хронической почечной недостаточностью (скорость клубочковой фильтрации <50 мл/мин). В рекомендациях предложены варианты лечения выделенных подгрупп пациентов, при этом особый акцент сделан на безопасность лечения пациентов с коморбидностью.
Исследования последних лет в области ОА позволили изменить отношение к этому заболеванию, которое долгое время считалось «Золушкой ревматологии» [6], так как не уделялось должного внимания этиопатогенетическим механизмам развития данного страдания, а следовательно, не разрабатывались новые методы лечения и диагностики. Современное видение ОА должно охватывать не только клиническую симптоматику, выявленную у пациента, но и степень структурных изменений суставных структур — суставного хряща, субхондральной кости, синовиальной оболочки, мышц, связок, менисков. Поскольку сустав воспринимается как орган, а ОА как заболевание, что поражает все суставные ткани, то возникает необходимость переоценки роли отдельных патогенетических факторов, оказывающих перекрестное комплексное влияние на сустав, а также их участие в прогрессировании заболевания. ОА сегодня требует системного подхода с учетом открытий молекулярной биологии, раскрытия этиопатогенетических механизмов инициации и прогрессирования болезни, формирования различных фенотипов течения ОА. Достижения в области визуализации и протеомики являются важным шагом в понимании патофизиологии ОА в свете меняющейся парадигмы заболевания. Неоднородность ОА подводит нас к пониманию высокой частоты неэффективного лечения этого заболевания, поскольку пациенты с различными фенотипами болезни и разной степенью прогрессирования требуют различных медикаментозных и немедикаментозных подходов. Поэтому чрезвычайно важна ранняя стратификация пациентов на различные клинические и патогенетические фенотипы, особенно на ранних стадиях заболевания, что подарит надежду и пациенту, и врачу на более успешное лечение.
Список использованной литературы
- Головач И.Ю. (2014) Остеоартрит: перезагрузка взглядов на патогенез заболевания. Что важно для реальной клинической практики? Здоровье Украины, 1(32): 66–68.
- Дубиков А.И. (2013) Остеоартроз: старая болезнь, новые подходы. Совр. ревматология, 2: 82–88.
- Зайцева Е.М., Алексеева Л.И., Насонов Е.Л. (2013) Патогенез остеоартроза и обоснование применения стронция ранелата. Науч.-практ. ревматология, 6: 696–702.
- Коваленко В.М., Борткевич О.П. (2010) Остеоартроз. Практична настанова — 3-тє вид., доп., зі змінами. МОРІОН, Киев, 608 с.
- Мендель О.И., Наумов А.В., Вёрткин А.Л. и др. (2010) Остеоартроз и сердечно-сосудистые заболевания у лиц пожилого возраста: клинические и патогенетические взаимосвязи. Успехи геронтологии, 23(2): 304–313.
- Хитров Н.А. (2013) Остеоартроз и остеоартрит — от новых взглядов на патогенез к новому названию. Мед. совет, 4: 74–78.
- Шуба Н.М., Воронова Т.Д., Тарасенко Т.М. та ін. (2012) Нові аспекти патогенезу остеоартрозу та шляхи його корекції. Укр. мед. часопис, 2(88): 113–119.
- Шумада И.В. (1990) Диагностика и лечение дегенеративно-дистрофических поражений суставов. Здоров’я, Київ, 196 с.
- Aigner T., Söder S., Gebhard P.M. et al. (2007) Mechanisms of Disease: role of chondrocytes in the pathogenesis of osteoarthritis — structure, chaos and senescence. Nat. Clin. Pract. Rheum., 3: 391–399.
- Berenbaum F. (2004) Signaling transduction: target in osteoarthritis. Curr. Opin. Rheumatol., 16(5): 616–622.
- Berenbaum F. (2011) Diabetes-induced osteoarthritis: from a new paradigm to a new phenotype. Ann. Rheum. Dis., 70: 1354–1356.
- Berenbaum F. (2013) Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage, 21: 16–21.
- Bijlsma J.W., Berenbaum F., Lafeber F.P. (2011) Osteoarthritis: an update with relevance for clinical practice. Lancet, 377: 2115–2126.
- Blagojevic M., Jinks C., Jeffery A. et al. (2010) Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta-analysis. Osteoarthritis Cartilage, 18: 24–33.
- Bougault C., Gosset M., Houard X. et al. (2012) Stress-induced cartilage degradation does not depend on NLRP3 inflammasome in osteoarthritis. Arthritis Rheum., 64(12): 3972–3981.
- Case J.P., Baliunas A.J., Block J.A. (2003) Lack of efficacy of acetaminophen in treating symptomatic knee osteoarthritis: a randomized, double-blind, placebo-controlled comparison trial with diclofenac sodium. Arch. Intern. Med.,163: 169–178.
- Castañeda S., Roman-Blas J.A., Largo R. et al. (2014) Osteoarthritis: a progressive disease with changing phenotypes. Rheumatology (Oxford), 53(1): 1–3.
- Catterrall J.B., Carrere S., Koshy P.J.T. et al. (2001) Synergic induction of matrix metalloproteinase 1 by interleukin-1α and oncostatin M in human chondrocytes involves signal transducer and activator of transcription factors via a novel mechanism. Arthritis Rheum., 44(10): 2296–2310.
- Dahlberg L., Billinghurst R.C., Manner P. et al. (2000) Selective enhancement of collagenase-mediated cleavage of resident type II collagen in cultured osteoarthritic cartilage and arrest with a synthetic inhibitor that spares collagenase 1 (matrix metalloproteinase 1). Arthritis Rheum., 43(3): 673–682.
- Denoble A.E., Huffman K.M., Stabler T.V. et al. (2011) Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proc. Natl. Acad. Sci USA, 108(5): 2088–2093.
- Esposito K., Nappo F., Marfella R. et al. (2002) Infl ammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation, 106: 2067–2072.
- Fernandes J.C., Martel-Pelletier J., Lascau-Coman V. et al. (1998) Collagenase-1 and collagenase-3 synthesis in normal and early experimental osteoarthritic canine cartilage: an immunohistochemical study. J. Rheumatol., 25(8): 1585–1594.
- Guilak F. (2011) Biomechanical factors in osteoarthritis. Best Pract. Res. Clin. Rheumatol., 25(6): 815–823.
- Knoop J., van der Leeden M., Thorstensson C.A. et al. (2011) Identification of phenotypes with different clinical outcomes in knee osteoarthritis: data from the Osteoarthritis Initiative. Arthritis Care Res., 63: 1535–1542.
- Loeser R.F., Goldrina S.R., Scanzello C.R. et al. (2012) Osteoarthritis: A Disease of the Joint as an Organ. Arthritis Rheum., 64(6): 1697–1707
- Lohmander L.S., Hoerrner L.A., Lark M.W. (1993) Metalloproteinases, tissue inhibitor, and proteoglycan fragments in knee synovial fluid in human osteoarthritis. Arthritis Rheum., 36(2): 181–189.
- Manicort D., Fujimoto N., Obata K. et al. (1994) Serum levels of collagenase, stromelysin-1, and TIMP-1. Arthritis Rheum., 37(12): 1774–1783.
- Martinon F., Pétrilli V., Mayor A. et al. (2006) Goutassociated uric acid crystals activate the NALP3 inflammasome. Nature, 440(7081): 237–241.
- McAlindon T.E., Bannuru R.R., Sullivan M.C. et al. (2014) OARSI guidelines for the non-surgical management of knee osteoarthritis. Osteoarthritis Cartilage, 22(3): 363–388.
- Nair A., Kanda V., Bush-Joseph C. et al. (2012) Synovial fluid from patients with early osteoarthritis modulates fibroblast-like synoviocyte responses to toll-like receptor 4 and toll-like receptor 2 ligands via soluble CD14. Arthritis Rheum., 64(7): 2268–2277.
- Pearle A.D., Scanzello C.R., George S. et al. (2007) Elevated high-sensitivity C-reactive protein levels are associated with local inflammatory findings in patients with osteoarthritis. Osteoarthritis Cartilage., 15 (5); 516–523.
- Roemer F.W., Guermazi A., Felson D.T. et al. (2011) Presence of MRI-detected joint effusion and synovitis increases the risk of cartilage loss in knees without osteoarthritis at 30-month follow-up: the MOST study. Ann. Rheum. Dis., 70(10): 1804–1809.
- Rosenthal A.K. (2011) Crystals, inflammation, and osteoarthritis. Curr. Opin. Rheumatol., 23(2): 170–173.
- Sanchez C., Pesesse L., Gabay O. et al. (2012) Regulation of subchondral bone osteoblast metabolism by cyclic compression. Arthritis Rheum., 64(4): 1193–1203.
- Scanzello C.R., McKeon B., Swaim B.H. et al. (2011) Synovial inflammation in patients undergoing arthroscopic meniscectomy: molecular characterization and relationship to symptoms. Arthritis Rheum., 63(2): 391–400.
- Scanzello C.R., Umoh E., Pessler F. et al. (2009) Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthritis Cartilage, 17(8): 1040–1048.
- Sellam J., Berenbaum F. (2010) The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol., 6 (11): 625–635.
- Spector T.D., Hart D.J., Nandra D. et al. (1997) Low-level increases in serum C-reactive protein are present in early osteoarthritis of the knee and predict progressive disease. Arthritis Rheum., 40: 723–727.
- Stevens A.L., Wishnok J.S., White F.M. et al. (2009) Mechanical injury and cytokines cause loss of cartilage integrity and upregulate proteins associated with catabolism, immunity, inflammation, and repair. Mol. Cell Proteomics, 8(7): 1475–1489.
- Sturmer T., Brenner H., Koenig W. et al. (2004) Severity and extent of osteoarthritis and low grade systemic inflammation as assessed by high sensitivity C reactive protein. Ann. Rheum. Dis., 63(2): 200–205.
- van Sloten T.T., Savelberg H.H., Duimel-Peeters I.G. et al. (2011) Peripheral neuropathy, decreased muscle strength and obesity are strongly associated with walking in persons with type 2 diabetes without manifest mobility limitations. Diabetes Res. Clin. Pract., 91: 32–39.
- Zhuo Q., Yang W., Chen J. et al. (2012) Metabolic syndrome meets osteoarthritis. Nat. Rev. Rheumatol., 8: 729–737.
Адрес для переписки:
Головач Ирина Юрьевна
03680, Киев, ул. Академика Заболотного, 21
Клиническая больница «Феофания» ГУД
E-mail: golovachirina@yandex.ru
Leave a comment