АНТИФОСФОЛІПІДНИЙ СИНДРОМ ПРИ НАБУТОМУ ДЕФІЦИТІ ФАКТОРІВ ЗГОРТАННЯ КРОВІ ТА ГЕНЕТИЧНО ЗУМОВЛЕНІЙ СХИЛЬНОСТІ ДО ТРОМБОТИЧНИХ ПОДІЙ У ДИТЯЧОМУ ВІЦІ
Муквіч О.М., Омельченко Л.І., Дяченко Н.М., Грідіна Т.А., Яковенко А.О.
Резюме. Антифосфоліпідний синдром (АФС) у дітей — потенційно небезпечне для життя аутоімунне мультисистемне захворювання, яке в основному проявляється тромбозом судин з високим ризиком повторного тромбозу (20–30%) і високим рівнем смертності (7%). Хоча тромбози у дітей відмічають набагато рідше, ніж у дорослих, частка тромбозів, пов’язаних з антифосфоліпідними антитілами, вища саме в педіатричній практиці. Мета дослідження: ознайомити лікарів з особливостями клінічних проявів та діагностики первинного АФС у дітей із гематологічними ускладненнями, важливістю проведення розширеного обстеження, включаючи дослідження всіх ланок гемостазу. Клінічний випадок. Наведено клінічний випадок власного спостереження первинного АФС у дитини з дебютом у підлітковому віці, посттромботичною хворобою, вираженими лабораторними ознаками глибокого порушення різних ланок гемостазу. Висновки. АФС є рідкісним розладом із обмеженою кількістю зареєстрованих випадків у дітей, але з різноманітними клінічними проявами і наслідками захворювання. Проведення розширеного обстеження дитини з АФС, включаючи дослідження всіх ланок гемостазу, дозволяє правильно обрати тактику лікування та спостереження, визначити заходи з профілактики тяжких ускладнень.
DOI:10.32471/rheumatology.2707-6970.95.18549
УДК 616-005.6-053.2:616.155.2-056.7
ВСТУП
Антифосфоліпідний синдром (АФС) — системне аутоімунне захворювання нез’ясованої етіології з широким спектром судинних та/або акушерських проявів, пов’язаних із тромботичними та запальними механізмами, керованими антифосфоліпідними антитілами (АФА) [10]. Поширеність АФС у педіатричній популяції України на сьогодні не визначена. За даними Канадського реєстру, в якому проспективно ідентифіковано 137 дітей, частота випадків венозного тромбозу становила 5,3 на 10 000 госпіталізацій, або 0,07 на 10 000 дитячого населення [1].
У патогенезі АФС провідну роль відіграють АФА, зокрема, анти-β2-глікопротеїн I (анти-β2-ГП І) антитіла, які являють собою різноманітну групу антитіл, що спрямовані проти ряду антигенних мішеней, таких як протромбін (ПТ), комплекси фосфатидилсерин/протромбін (ФС/ПТ), комплекси віментин/кардіоліпін, аннексин A2 і аннексин A5. АФА-індукована клітинна активація призводить до множинних прокоагулянтних і прозапальних ефектів, а також активації комплементу і є основним чинником тромботичних ускладнень при АФС. Наявність додаткових факторів ризику, таких як системний червоний вовчак (СЧВ), спадкова тромбофілія, злоякісні новоутворення, куріння, вагітність та застосування оральних контрацептивів, що містять естроген, додатково підвищує ризик тромбоутворення у пацієнтів з АФА [12].
Розрізняють первинний та вторинний АФС, який виникає при його асоціації з іншим аутоімунним захворюванням (СЧВ, ювенільний ідіопатичний артрит та ін.). У дітей може також розвиватися катастрофічний АФС, останній характеризується швидким розвитком поліорганного тромбозу та мікроангіопатією дрібних судин, рідше виникає тромбоз великих судин [6, 9].
Оновлені критерії класифікації АФС за стандартами Європейського альянсу ревматологічних асоціацій (European Alliance of Associations for Rheumatology — EULAR) 2023 у дорослих [3] включають клінічні та лабораторні показники (таблиця).
Таблиця. Класифікаційні критерії АФС American College of Rheumatology (ACR)/EULAR 2023 р.
| Вхідний критерій≥1 задокументованого клінічного критерію + ≥1 позитивного АФА (протягом 3 років після клінічного критерію) | |||
|---|---|---|---|
| Клінічний домен | Бал | Лабораторний домен | Бал |
| Венозна тромбоемболія (ВТЕ) | Вовчаковий антикоагулянт | ||
| Високий ризик ВТЕ | 1 | Виявлений одноразово | 1 |
| Без високого ризику ВТЕ | 3 | Персистуючий | 5 |
| Артеріальні тромбози | Антикардіоліпінові антитіла (аКЛ)/анти-β2-глікопротеїни (анти-β2-ГП І) (ELISA 40-79 ОД — помірний, ≥80 ОД — високий) | ||
| Високий кардіоваскулярний ризик (КВР) | 2 | Помірний/високий рівень аКЛ та/або анти-β2-ГП І IgM | 1 |
| Без високого КВР | 4 | Помірний рівень аКЛ та/або анти-β2-ГП І | 4 |
| Мікроваскулярне залучення* | Високий рівень аКЛ або анти-β2-ГП І | 5 | |
| Підозра | 2 | Високий рівень аКЛ та анти-β2-ГП І IgG | 7 |
| Встановлене | 5 | ||
| Акушерський |
Зараховується тільки найбільший критерій в кожному домені. Не зараховується, якщо є більш очевидне роз’яснення. *Мікроваскулярне залучення Підозра: ліведо racemosа, лівеоїдна васкулопатія, АФА-нефропатія, легенева кровотеча (симптоми, візуалізація). Встановлено: лівеоїдна васкулопатія (біопсія), АФА-нефропатія (біопсія), легенева кровотеча (бронхоальвеолярний лаваж, біопсія), інфаркт міокарда/надниркова кровотеча (гістологія або візуалізація). |
||
| ≥3 послідовних втрат (≤10 тиж) та/або смерть плода (≥10–15 тиж, 6 днів) | 1 | ||
| Смерть плода (16–33 тиж, 6 днів) без прееклампсії (ПЕ)/плацентарної недостатності (ПН) або ПН з тяжкими ознаками | 1 | ||
| Тяжка ПЕ або ПН (<34 тиж) з або без смерті плода | 3 | ||
| Тяжка ПЕ та ПН (<34 тиж) з або без смерті плода | 4 | ||
| Клапани серця | |||
| Потовщення | 2 | ||
| Вегетації | 4 | ||
| Тромбоцитопенія (20–130 г/л) | 2 | ||
|
Загальний підрахунок Класифікувати як АФС, якщо є принаймні 3 бали з клінічних ділянок та принаймні 3 бали з лабораторних показників |
|||
Залежно від ураженої системи органів клінічні некритеріальні прояви АФС поділяють на декілька підгруп: серцево-судинні, неврологічні, шкірні, ниркові, гематологічні та інші. Гематологічні прояви включають тромбоцитопенію, гемолітичну анемію та функціональні зміни або дефіцит факторів згортання крові з тенденцією до тромбозу (набута резистентність до активованого протеїну С, дефіцит протеїну S) або кровотечі [2]. Зв’язок АФА з тромбоемболічними подіями широко та добре задокументований. Однак набута коагулопатія, спричинена АФА, є комплексною й іноді може проявлятися у вигляді геморагічної події з різним клінічним ступенем тяжкості або комбінованого тромбогеморагічного синдрому [7, 11].
При венозному тромбозі та стійко позитивному тесті на АФА поточна рекомендація полягає в лікуванні довготривалими антикоагулянтами [15].
Лікування АФС у дітей на сьогодні базується на модифікованих рекомендаціях лікування АФС у дорослих, але через вікові особливості гемостазу у дітей не може повною мірою відповідати їх терапевтичним потребам. У АФА-позитивних дітей використовується гідроксихлорохін, який також може забезпечити деякі додаткові можливості для профілактики ускладнень АФС [4].
Враховуючи вищевикладене, на сьогодні немає чітких рекомендацій, що ґрунтуються на доказах, відносно діагностики та лікування АФС у дітей, що потребує додаткових досліджень, в тому числі аналізу окремих клінічних випадків [14].
МЕТА РОБОТИ
Ознайомити лікарів з особливостями клінічних проявів та діагностики первинного АФС у дітей із гематологічними ускладненнями, важливістю проведення розширеного обстеження, включаючи дослідження всіх ланок гемостазу.
Наводимо клінічний випадок власного спостереження первинного АФС у дитини з дебютом у препубертатному віці, посттромботичною хворобою, вираженими лабораторними ознаками глибокого ураження ланок гемостазу.
Хлопчик віком 17 років вперше госпіталізований у відділення дитячої ревматології та аутозапальних захворювань ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України» через 8 років від початку захворювання зі скаргами на звивистість та напруження вен правої гомілки, набряк її та біль переважно у вечірній час, висип в підколінній ділянці правої ноги, кровоточивість ясен під час чищення зубів.
У віці 9 років під час спортивних змагань (гра у баскетбол) отримав травму з утворенням у верхній частині правої гомілки гіперемованої ущільненої ділянки, гарячої на дотик, різко болючої при пальпації та обмеженням рухів у правому колінному суглобі. Після обстеження в обласній лікарні за даними ультразвукового дослідження (УЗД) судин нижніх кінцівок діагностовано флеботромбоз вен правої гомілки (проходив стаціонарне лікування, яке включало інфузійну терапію антикоагулянтами). У подальшому спостерігався судинним хірургом 2 рази на рік, періодично отримував медикаментозну терапію (детралекс, варфарин, фраксипарин, тивортин, актовегін протягом одного року, далі 5 років отримував цикло-3 форт, нормовен). В останні 1,5–2 роки лікарські засоби не отримував. У віці 14 років вперше виявлено патологічні зміни коагулограми: підвищення рівня активованого часткового тромбопластинового часу (АЧТЧ) — 43,3с (N 22–33), час рекальцифікації згустку — 1,72 с (N 0,8–1,2), підвищення рівня АФА: скринінговий тест на вовчаковий антикоагулянт — 106,2 с (N 31–44), підтверджувальний тест — 42,8 с (N 30–38), LA-AUTO — 2,4813 (сильно виражений ризик). Одночасно виявлені підвищені рівні аКЛ IgG >160 (сильно позитивний результат) та β2-глікопротеїду І антитіл IgG >160 (сильно позитивний результат). Дитячим ревматологом амбулаторно встановлено діагноз АФС, призначене лікування пацієнт не отримував. При повторних обстеженнях в медичних установах України утримувалися підвищені рівні АЧТЧ (55,1–76,9 с), подовження протромбінового часу (20,2–23,1 с), протромбін за Квіком 53%.
У віці 17 років при обстеженні в центрі гемостазу НДСЛ «ОХМАТДИТ» вперше виявлено дефіцит VIII (7,8%), IX (2,2%), XI (3,8%) факторів згортання крові. При цьому морфологія тромбоцитів (макроформи — 2%, мезоформи — 82%, мікроформи — 16%) та показники агрегації тромбоцитів з аденозиндифосфатом (АДФ) і ристоцетином мали нормативні значення, а з епінефрином та колагеном виявлено зниження активності тромбоцитів. За даними біохімічного аналізу крові, виявлено підвищений рівень гомоцистеїну — 19,3 ммоль/л (N до 12,4) при нормативних показниках фолієвої кислоти — 1,85 нг/мл (N 1,2–8,8) та вітаміну В12 — 282,3 пг/мл (N 214–864). За даними інструментальних досліджень, виявлено ехографічні ознаки гепатоспленомегалії, ультразвукові симптоми посттромбофлебітичного синдрому, кіста Бейкера лівої підколінної ділянки.
При проведенні молекулярно-генетичного дослідження в ДЗ «Референс-центр з молекулярної діагностики МОЗ України» встановлена експресія генів PAI-I, FGB, ITGA2a, Factor XII, HpaA-2b, MTHFR, MTRR, MTRl, RFC, що дозволило діагностувати спадкову гіпергомоцистеїнемію помірного ступеня, змішаного ґенезу, зумовлену варіантами генів фолатного обміну MTHFR, MTRR, MTR1, RFC та коагулопатію (глибокий дефіцит факторів VІІІ, ІХ, ХІ), низький фолатний статус. Діагностовано генетично зумовлену схильність до тромбофілічних розладів, асоційовану з варіантами генів PAI-I, FGB, ITGA2α, Factor XII, HpaA-2b. Зниження чутливості до фолієвої кислоти, 5-метилтетрагідрофолату, ціанокобаламіну; високий ризик множинних гіповітамінозних станів, анемії, неалкогольної жирової дистрофії печінки; уповільнене накопичення та виведення ліків, ендогенних шкідливих сполук та екзогенних метаболітів, асоційоване із варіантом гена MDR1. Підозра на спадкове порушення обміну амінокислот та ацилкарнітинів.
Для уточнення діагнозу та визначення подальшої тактики терапії дитина направлена в ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України».
З анамнезу життя відомо, що дитина від І вагітності, ускладненої поєднаним гестозом на тлі гіпертонічної хвороби матері (перебувала на стаціонарному лікуванні), І термінових пологів на 38-му тижні шляхом кесаревого розтину (внутрішньоутробна гіпоксія плода, туге дворазове обвиття пуповиною). Неонатальний період не супроводжувався геморагічними епізодами. Прорізування зубів, фізична активність не супроводжувалися гематомами, синцями. Серед дитячих інфекційних захворювань — вітряна віспа в 5 років. Хворіє на гострі респіраторні інфекції 3–4 рази на рік. Щеплений за віком, без ускладнень. Серед травм: перелом правої променевої кістки у віці 4 роки, в 13 років травма губи, проведена механічна зупинка гемостазу (накладання швів). Оперативне втручання з приводу пупкової кили у віці 6 років без геморагічних ускладнень. За даними релевантного анамнезу, алергологічних проявів не визначено, спадковість обтяжена (у мами та бабусі за материнською лінією варикоз вен нижніх кінцівок, артеріальна гіпертензія).
На час госпіталізації у відділення температура тіла 36,6 ˚С, частота серцевих скорочень (ЧСС) 75 уд./хв, маса тіла 75 кг, зріст 193 см, індекс маси тіла (ІМТ) = 20,1, S=2,04 м2. Загальний стан дитини стабільний. На внутрішній поверхні правого стегна в підколінній ділянці обох ніг візуалізується висип у вигляді червоно-коричневих плям овальної форми із лущенням, на гомілках — поодинокі синці, посттравматичні рубці. На задній поверхні правої гомілки вени звивисті, при пальпації еластичної консистенції, підвищеного напруження. Видимі слизові оболонки рожеві, вологі. Нігтьові пластини не уражені, волосся не випадає. Язик рожевий, обкладений білим нальотом, зуби сановані, ясна періодично кровоточать. Периферичні лімфатичні вузли шийної групи збільшені до 0,5 см, еластичні, безболісні, не спаяні з навколишніми тканинами, поодинокі. М’язовий тонус симетричний, збережений. Перкуторно відмічається ясний легеневий звук. Аускультативно дихання везикулярне, хрипи не вислуховуються. Тони серця приглушені, ритмічні. Живіт м’який, безболісний, доступний для поверхневої пальпації. Печінка біля краю реберної дуги, селезінка не пальпується. Діурез достатній, дефекація 1 раз на добу, випорожнення забарвлені, без патологічних домішок. Суглоби не дефігуровані, не гарячі, не набряклі, безболісні при пальпації, активні та пасивні рухи в повному обсязі, функція збережена. Болісність остистих відростків відсутня, обмеженості рухів у шийному відділі хребта не виявлено.
Під час перебування в стаціонарі проводили дослідження для виключення вторинного АФС, асоційованого із СЧВ (антинуклеарні антитіла <1:100 (негативні), SmDP-S, антитіла IgG — 1 Од/мл (негативні), комплемент (С3С компонент) — 0,93 г/л (N 0,9–1,8), комплемент (С4 компонент) — 0,15 г/л (N 0,1–0,4), антитіла до дволанцюгової ДНК — 0,8 (N <1,1). Лабораторно на тлі різко підвищених рівнів АФА: αCL IgG — 54,1 GPL (N <9), αPS IgG — 161,4 U/ml (N <9), αPE IgG — 58 U/ml (N <10) утримувалося підвищення АЧТЧ до 128,2 с, протромбінового часу — до 20,1 с. При повторному обстеженні в центрі гемостазу виявлено негативну динаміку окремих показників згортання крові: фактор ІІ — 31,6% (N 79–131), фактор V — 65,2% (N 62–139), фактор VII — 81,7% (N 50–129), фактор VIIІ — 5,5% (N 50–150), фактор ІХ — 0,0% (N 65–150), фактор Х — 93,7% (N 77–131), фактор ХІ — 0,0% (N 65–150), фактор ХІІ — 0,0% (N 50–150), фактор фон Віллебранда (Ag) — 99,7% (N 61–157), фактор фон Віллебранда (active) — н/в (N 49–169), протеїн С — 94,8% (N 70–140), протеїн S, вільний — 104,4% (N 57,6–112,5). Агрегація тромбоцитів з АДФ і епінефрином знижена, з ристоцетином відсутня.
При УЗД діагностовано гепатоспленомегалію, варикозне розширення селезінкової вени (рис. 1), формування портальної гіпертензії, що розцінено як початкові ознаки формування синдрому Бадда — Кіарі.
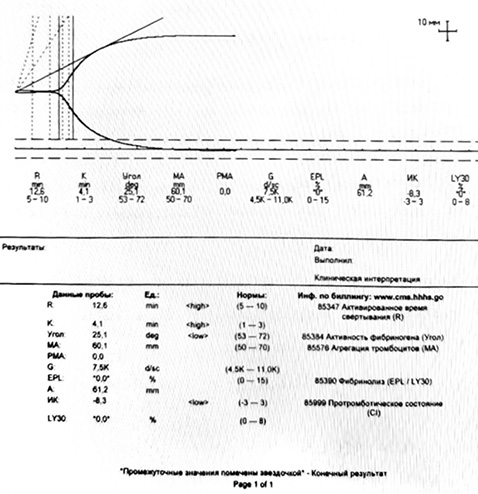
За даними допплерометрії судин нижніх кінцівок: права н/к: клубові вени та загальна стегнова вена (ЗСВ) прохідні, субтотальна реканалізація поверхневих стегнових вен (ПСВ) та підколінно-гомілкового сегмента, рефлюкс крові. Рефлюкс за верхньою порожнистою веною (ВПВ). Клапани малої підшкірної вени (МПВ) спроможні. Недостатність поверхневих вен (ПВ) Cockett, Dodd, задньо-медіальної; ліва н/к: клубові вени та ЗСВ прохідні. Субтотальна реканалізація ПСВ та підколінно-гомілкового сегмента, рефлюкс крові. ВПВ та МПВ прохідні, клапани спроможні. При проведенні тромбоеластографії показники гемостазу вказують на стан гіпокоагуляції (рис. 2).
На підставі скарг, клініко-анамнестичних даних, лабораторних та інструментальних досліджень, проведеної диференційної діагностики дитині встановлено діагноз первинний АФС (позитивні антитіла IgG до кардіоліпіну, β2-глікопротеїду, антифосфатидилсеринові, антифосфатидилетаноламінові, вовчаковий антикоагулянт; варикозне розширення селезінкової вени, в анамнезі 2 епізоди флеботромбозу глибоких вен нижніх кінцівок). Портальна гіпертензія, синдром Бадда — Кіарі? Генетично зумовлена схильність до тромбофілічних розладів, асоційована із варіантами генів PAI-I, FGB, ITGA2α, Factor XII, HpaA-2b. Вторинна коагулопатія (глибокий дефіцит факторів VIII, IX, XI, XII). Посттромботична хвороба обох нижніх кінцівок. Вторинний варикоз правої нижньої кінцівки. Хронічна венозна недостатність правої нижньої кінцівки С3, лівої — С2 (СЕАР). Порушення фолатного обміну.
Дитині призначено терапію гідроксихлорохіном та ацетилсаліциловою кислотою для профілактики та попередження повторних випадків тромбозу, носіння компресійних панчох ІІІ класу на нижні кінцівки в денний час. Заплановано оперативне лікування варикозно розширених вен нижніх кінцівок — ендовенозна лазерна коагуляція (ЕВЛК).
ОБГОВОРЕННЯ
Представлений клінічний випадок первинного АФС у дитини віком 17 років з необтяженим сімейним анамнезом демонструє його пізню діагностику, оскільки дебют захворювання, очевидно, мав місце в препубертатному віці, коли у дитини у віці 9 років після спортивної травми був виявлений субклінічний тромбофлебіт вен травмованої гомілки. Хоча дитині було проведено стаціонарне лікування антикоагулянтами та судинними препаратами та рекомендовано їх прийом в амбулаторних умовах, діагноз АФС вперше було встановлено лише у віці 14 років, коли в сироватці крові виявлено патологічні зміни коагулограми та високі рівні АФА разом з вовчаковим антикоагулянтом.
Особливістю випадку є розвиток у пацієнта рідкісного гематологічного ускладнення при АФС, вторинної коагулопатії (гіпокоагуляції), спричиненої набутим глибоким дефіцитом факторів VIII, IX, XI, XII на тлі генетично зумовленої схильності до тромбофілічних розладів, асоційованих з варіантами генів PAI-I, FGB, ITGA2α, Factor XII, HpaA-2b.
Інгібітори факторів згортання крові — це антитіла, які нейтралізують активність факторів згортання крові та підвищують їх кліренс, перешкоджаючи, тим самим, їх нормальній функції, як це відмічено при таких станах, як гемофілія, аутоімунні захворювання, злоякісні пухлини, вагітність та побічні ефекти від прийому препаратів. Вовчаковий антикоагулянт є антифосфоліпідним антитілом, яке пов’язане з ризиком тромбозу, як це відмічають при АФС. У рідкісних випадках у пацієнтів з наявністю вовчакового антикоагулянта можуть виникати геморагічні ускладнення [8].
У пацієнта глибокі порушення ланок гемостазу проявлялися повною відсутністю VIII, IX, XI, XII факторів згортання крові. Відомо, що фактор XI — це глікопротеїн плазми крові, який бере участь у ранній фазі каскаду згортання крові, виявляється при нормальному гемостазі і коливається в межах 70–150%. Тяжкий дефіцит фактора XI визначається як активність <15% від норми, а особи з частковим дефіцитом — зазвичай мають активність 20–70% відносно норми. Набутий дефіцит фактора XI часто виявляють у пацієнтів із захворюваннями печінки внаслідок недостатнього його синтезу або в період початку синдрому дисемінованого внутрішньосудинного згортання крові, а також у хворих зі злоякісними захворюваннями та при аутоімунних хворобах, зокрема СЧВ [5].
Формування аутоантитіл проти факторів згортання крові відбувається у 10–15% АФА-позитивних пацієнтів. Зазвичай АФС являє собою набуту аутоімунну тромбофілію із тенденцією до гіперкоагуляції. У нашому випадку набутий дефіцит факторів згортання крові зумовив, з вищою вірогідністю, рух системи гемостазу в бік гіпокоагуляції.
Наведені особливості перебігу АФС у пацієнта визначили необхідність персоніфікованого підходу до вибору терапевтичної тактики з огляду на ризики кровотечі та тромбоутворення. Пацієнту було призначено гідроксихлорохін, оскільки в in vivo, in vitro та клінічних дослідженнях продемонстрована його ефективність щодо зниження ризику тромбозу [13]. Враховано також, що гідроксихлорохін не пов’язаний із підвищеним ризиком кровотечі при застосуванні його як в монотерапії, так і в комбінації з антикоагулянтами [4].
Враховуючи, що у пацієнта є глибоке порушення гемостазу з відсутністю окремих факторів згортання крові, а в літературі — повідомлення про можливі кровотечі при застосуванні атикоагулянтів, ці препарати хворому не рекомендували. Призначено антитромботичні препарати, а саме ацетилсаліцилову кислоту в дозі 75 мг на добу. Один з метааналізів з 11 досліджень, включно з 1208 пацієнтами з АФА і 139 тромботичними подіями, свідчить, що пацієнти з АФА, які довго отримували низькі дози ацетилсаліцилової кислоти, мали на 50% нижчий ризик виникнення тромботичної події, ніж ті, хто не отримував цього лікування [12]. Крім того, аналіз підгруп відповідно до патогенетичного фону показав, що зниження ризику було значним у безсимптомних АФА-позитивних осіб, а також у АФА-позитивних пацієнтів із СЧВ або тромботичним АФС.
ВИСНОВКИ
Наведений клінічний випадок свідчить, що первинний АФС у дітей є тяжким для діагностики та лікування захворюванням, що загрожує життю або знижує його якість. У дитячому віці для його виявлення необхідно оцінювати наявність не лише класифікаційних критеріїв та некритеріальних ускладнень з боку серцево-судинної, нервової систем, нирок, печінки, шкіри та ін., а й проведення ретельного лабораторного обстеження для виявлення коагулопатій. Важливим у діагностиці та оцінці ризику повторних тромбозів є генетичне обстеження на наявність вродженої схильності до тромботичних розладів.
Діти з АФС потребують персоніфікованого підходу до лікування, при цьому рішення щодо застосування антикоагулянтів при вторинній профілактиці тромбозу має бути обґрунтоване розширеним обстеженням дитини з АФС, включаючи проведення дослідження всіх ланок гемостазу. Довічна антикоагулянтна терапія далека від ідеалу, тому необхідні більш точні інструменти для визначення ризику та прогнозу. Проблема АФС у дітей потребує подальших досліджень для створення рекомендацій з діагностики, лікування захворювання та схеми спостереження таких дітей, оскільки при відповідних ліках і модифікації способу життя більшість дітей з первинним АФС можуть вести нормальний здоровий спосіб життя.
Залежно від локалізації та ступеня тромбозу рідкісне гематологічне ускладнення у дитини з первинним АФС у вигляді формування вторинної коагулопатії (глибокий дефіцит VIII, IX, XI, XII факторів згортання крові) із поєднанням з генетично зумовленою схильністю до тромбофілічних розладів із варіантами генів PAI-I, FGB, ITGA2a, Factor XII, HpaA-2b може призводити як до геморагічного діатезу, так і до ризику тромбозу. Для відповідного лікування необхідна ретельна інтерпретація результатів лабораторних аналізів за наявності АФА. Більш високий ризик повторного тромбозу пов’язаний з наявністю позитивного результату на вовчаковий антикоагулянт та/або наявністю високих титрів АФА (тобто позитивний на вовчаковий антикоагулянт, антикардіоліпінові та антитіла до β2-глікопротеїну I), на відміну від позитивного результату лише одного тесту. У АФА-позитивних дітей необхідно досліджувати некритеріальні ускладнення, такі як серцево-судинні, неврологічні, шкірні, ниркові, гематологічні для якісного підбору терапії та рекомендацій щодо подальшого спостереження.
СПИСОК ВИКОРИСТАНОЇ ЛІТЕРАТУРИ
- 1. Дудник В.М., Фурман В.Г., Демянишина В.В. (2018) Антифосфоліпідний синдром у дітей. Вінницький національний ме-дичний університет імені М.І. Пирогова, Україна.
- 2. Abreu M.M., Danowski A., Wahl D.G. et al. (2015) The relevance of «non-criteria» clinical manifestations of antiphospholipid syndrome: 14th international congress on antiphospholipid antibodies. Autoimmun. Rev.;14: 401–414. DOI: 10.1016/j.autrev.2015.01.002.
- 3. Barbhaiya M., Zuily S., Naden R. et al. (2023) The 2023 ACR/ EULAR Antiphospholipid Syndrome Classification Criteria. Arthritis Rheumatol. Oct; 75(10): 1687–1702. doi: 10.1002/art.42624. Epub 2023 Aug 28. PMID: 37635643.
- 4. Belizna C. (2015) Hydroxychloroquine as an anti-thrombotic in antiphospholipid syndrome, Autoimmunity Reviews, Volume 14, Issue 4: 358–362. ISSN 1568-9972, doi.org/10.1016/j.autrev.2014.12.006.
- 5. Bolton-Maggs P.H., Patterson D.A., Wensley R.T. et al. (1995) Definition of the bleeding tendency in factor XI-deficient kind reds–a clinical and laboratory study. Thrombosis and Haemostasis, vol. 73, 2: 194–202.
- 6. Garcia D., Erkan D. (2018) The antiphospholipid syndrome. N. Engl. J. Med.; 378: 2010–2021. DOI: 10.1056/NEJMra1705454.
- 7. Kubisz P., Holly P., Stasko J. (2021) Bleeding in Patients with Antiphospholipid Antibodies. . Submitted: January 2nd, Reviewed: April 23rd, 2021. Published: June 4th, 2021. DOI: 10.5772/intechopen.97856.
- 8. Lim S., Zuha R., Burt T. et al. (2006) Life-threatening bleeding in a patient with a lupus inhibitor and probable acquired factor VII deficiency. Blood Coagulation & Fibrinolysis, vol.17, 8: 667–671.
- 9. Madison J.A., Zuo Y., Knight J.S. (2019) Pediatric antiphospholipid syndrome. Eur. J. Rheumatol., Dec 3; 7(Suppl. 1): 1–10. doi: 10.5152/eurjrheum.2019.19160. Epub ahead of print. PMID: 31804173; PMCID: PMC7004270.
- 10. Negrini S., Pappalardo F., Murdaca G. et al. (2017) The antiphospholipid syndrome: from pathophysiology to treatment. Clin. Exp. Med., 17: 257–267. doi.org/10.1007/s10238-016-0430-5.
- 11. Pazzola G., Zuily S., Erkan D. (2015) The challenge of bleeding in antiphospholipid antibody-positive patients. Curr. Rheumatol. Rep.; 17: 7. DOI: 10.1007/s11926-014-0481-0.
- 12. Pediatric Antiphospholipid Antibody Syndrome (2021) medscape.com Updated: Aug 25, URL: emedicine.medscape.com/ article/1006128-overview#a5.
- 13. Rand J.H., Wu X.X., Quinn A.S. et al. (2008) Hydroxychloroquine directly reduces the binding of antiphospholipid antibody-beta2-glycoprotein I complexes to phospholipid bilayers. Blood, Sep 1; 112(5): 1687–95. doi: 10.1182/blood-2008-03-144204. Epub 2008 Jun 24. PMID: 18577708; PMCID: PMC2518879.
- 14. Tektonidou M.G., Andreoli L., Limper M. et al. (2019) EULAR recommendations for the management of antiphospholipid syndrome in adults Annals of the Rheumatic Diseases; 78: 1296–1304. URL: ard.bmj.com/content/78/10/1296.
- 15. Zuily S., Cohen H., Isenberg D. et al. (2020) Use of direct oral anticoagulants in patients with thrombotic antiphospholipid syndrome: Guidance from the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. J. Thromb. Haemost.; 18: 2126– 2137. doi.org/10.1111/jth.14935.
Відомості про авторів:
Муквіч О.М., доктор медичних наук, професор, завідувачка відділення ревматичних хвороб у дітей ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України», м. Київ; керівник референтного центру з питань рідкісних захворювань за напрямами системні, ревматологічні хвороби дитячого віку МОЗ України.
E-mail: olena.mukvich@gmail.com.
ORCID ID: 0000-0001-6405-4997
Омельченко Л.І., доктор медичних наук, професор, головний науковий співробітник відділення ревматичних хвороб у дітей ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України», м. Київ.
ORCID ID: 0000-0003-2989-9278
Дяченко Н.М., дитячий кардіоревматолог, лікар-педіатр ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України», м. Київ.
ORCID ID: 0000-0003-3949-8857
Грідіна Т.А., кандидат медичних наук, лікар ультразвукової діагностики, ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України», м. Київ.
ORCID ID: 0000-0002-9234-9896
Яковенко А.О., лікар-педіатр ДУ «Інститут педіатрії, акушерства і гінекології імені академіка О.М. Лук’янової НАМН України», м. Київ.
ORCID ID: 0009-0007-5694-5328
Leave a comment