ЮВЕНІЛЬНИЙ ДЕРМАТОМІОЗИТ. ПРО ЩО ВАРТО ПАМ’ЯТАТИ ДОРОСЛОМУ РЕВМАТОЛОГУ?
Ошлянська О.А.1, Шевченко Н.С.2, Білявська Ю.В.3
- 1Національний університет охорони здоров’я України імені П.Л. Шупика, Київ
- 2Харківський національний університет ім. В.Н. Каразіна
- 3ДУ «ННЦ «Інститут кардіології імені акад. М.Д. Стражеска» НАМН України
Резюме. >Обґрунтування. Ювенільний дерматоміозит (ЮДМ) — тяжке рідкісне прогресуюче системне захворювання аутоімунного ґенезу з переважним ураженням поперечносмугастих м’язів, шкіри та судин мікроциркуляторного русла, яке розвивається у дітей до 16-річного віку, але незважаючи на тяжкість, смертність від ЮДМ значно нижча, ніж у пацієнтів дорослого віку з дерматоміозитом/поліміозитом, також меншою вдвічі є інвалідизація у пацієнтів дитячого віку. ЮДМ має певні імунологічні та клінічні відмінності розвитку та перебігу у дітей порівняно з особами дорослого віку. Методи дослідження. На підставі літературних даних у статті наведені сучасні уявлення щодо етіології та патогенезу, описані діагностичні критерії ЮДМ; проведене зіставлення клінічних та лабораторних проявів і частоти їх виявлення у пацієнтів дитячого та дорослого віку, підкреслені основні відмінності диференційної діагностики. Наведені уніфіковані терапевтичні стратегії відповідно до рекомендацій міжнародних ревматологічних асоціацій. Висновки. Враховуючи поточні терапевтичні можливості, пацієнти з ЮДМ становитимуть все більшу частку серед пацієнтів із запальними міозитами. Це обґрунтовує потребу у подальших дослідженнях менеджменту хворих із ЮДМ для покращення рівня надання медичної допомоги та віддалених наслідків хвороб.
DOI: 10.32471/rheumatology.2707-6970.86.16560
УДК: 616.5+616.74–002/-053.2
Вступ
Ювенільний дерматоміозит (ЮДМ) — тяжке рідкісне прогресуюче системне захворювання аутоімунного ґенезу з переважним ураженням поперечносмугастих м’язів, шкіри та судин мікроциркуляторного русла, яке розвивається у дітей віком до 16 років (США до 18 р.) [1].
Епідеміологія
Захворюваність на ЮДМ є остаточно не вивченою і коливається від 2,2 до 4,2 на 1 млн дітей, з яких менше 5% становлять випадки з ураженням лише м’язів (поліміозит), що значно менше, ніж у дорослому віці, в якому захворюваність у середньому становить 20 на 1 000 000 населення, а частота поліоміозиту сягає 29–65% [2]. Демографічні дослідження, проведені в США [3], відмічають етнічні відмінності захворюваності, яка є найбільшою для дітей європеоїдної раси та найменшою для осіб латиноамериканського походження. Поширеність ЮДМ також дуже низька — 4–6 на 100 тис. дитячого населення і є значно меншою, ніж така дерматоміозиту у дорослих (30 на 100 тис.). Загалом хворі на ЮДМ становлять лише 7% серед усіх випадків запальних міопатій. ЮДМ розвивається найчастіше у дітей молодшого віку, причому визначено два умовні піки дебюту ЮДМ: у 3–5 та у 7–9 років відповідно. На відміну від дорослих, ЮДМ відмічають частіше у дівчаток (2:1) [4], відрізняється віковий розподіл у дітей різної статі: середній вік діагнозу ЮДМ становить 6,7 року у дівчаток і 7,3 року у хлопчиків [5]. В певних регіонах (наприклад острівні держави, такі як Великобританія) виявлено ще більш виражені гендерні відмінності (5:1) [6]. Повідомляється, що співвідношення дівчаток і хлопчиків при ранньому дебюті (до 3 років) більш виражене (3,5:1 порівняно зі старшою групою 1,4:1) [7]. Але необхідно відзначити, що протягом останніх десятиліть, коли ведеться активне вивчення ЮДМ, показники захворюваності, поширеності та демографічні характеристики щодо віку дебюту та гендерного співвідношення суттєво не змінилися [3].
Важливо відмітити те, що смертність хворих на ЮДМ значно нижча, ніж у дорослих пацієнтів з дерматоміозитом/поліміозитом і становить менше 3%, також у пацієнтів дитячого віку вдвічі меншою є й інвалідизація (до 40%), що зумовлено удосконаленням підходів до виявлення і лікування захворювання в останні роки [8, 9].
Основи етіології та патогенезу
Як і всі захворювання аутоімунного ґенезу, ЮДМ не має чіткого етіологічного чинника. Незважаючи на численні повідомлення про виявлення методом електронної мікроскопії вірусоподібних частинок у м’язових волокнах хворих на ЮДМ, остаточно його вірусна етіологія ані серологічними, ані вірусологічними методами не підтверджена [10]. Здебільшого експерти вважають інфекції (коксакі-віруси, ентеровіруси, стрептококи, парвовірус В19) тригерними чинниками розвитку цієї аутоімунної хвороби у пацієнта з певною генетичною схильністю [11–20]. Слід підкреслити, що зазначені тригери ЮДМ (такі як вірусні інфекції та ультрафіолетове випромінення, особливо в регіонах, ближчих до екватора) також відрізняються від ініціюючих чинників дерматоміозиту у дорослих [16]. Крім того, доведено, що розвиток інфекційного синдрому в перші 6 міс від дебюту захворювання підвищує ризик рецидивного перебігу ЮДМ [21]. В якості одного з тригерних факторів ЮДМ розглядається і забруднення навколишнього середовища [18, 19].
У країнах, де відбувається зміна всіх сезонів (Росія, США), раніше (у 1980 р.) відмічена весняно-осіння сезонність народження дітей, у яких згодом розвивається ЮДМ, чого зараз не відмічають внаслідок глобального потепління [22]. До генетичних предикторів підвищеного ризику розвитку ЮДМ належать наявність HLA DRB1*0301-DQA1*0501, а також TNF-α -308A, PTP N22 R620W та Gm 3, 23, 5,13 алелів у осіб європеоїдної раси [23–26]. І навпаки, виявлення DQA1* 01 та DQA1 F25 можна розглядати як захисний фактор, у носіїв якого захворювання зазвичай не розвивається [24]. Відомо також, що найчастішими імуногенетичними маркерами ЮДМ є поєднання HLAB8 і DR3, тоді як для поліміозиту, асоційованого з системними хворобами сполучної тканини, більш характерне поєднання HLAВ14 і В40 [26].
Провідною ланкою патогенезу ЮДМ є розвиток порушень імунологічної толерантності та імуноопосередкованої васкулопатії судин дрібного калібру (рис. 1).
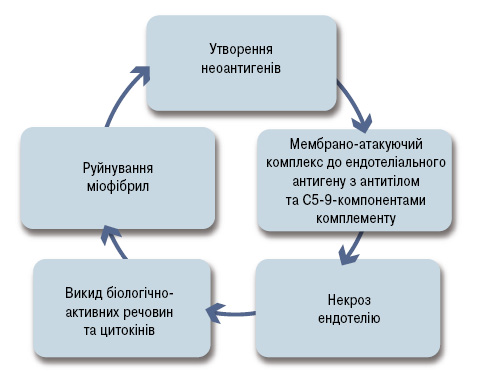
Виявлення у крові хворих на ЮДМ лімфоцитів, що реагують на антигени скелетних м’язів, а також наявність лімфоцитарної інфільтрації в уражених м’язах, свідчать про можливу участь у патогенезі хвороби клітинно-опосередкованої імунної реакції. Доведено, що у формуванні деструктивного васкуліту при ЮДМ активну участь беруть Т-лімфоцити-хелпери-1, -2 і -17, які інфільтрують м’язову тканину, стимулюють локальний синтез інтелейкіну-2 і фактору некрозу пухлин (ФНП), що ініціюють системну запальну відповідь [27].
В останні роки особлива увага вчених прикута до ролі дендритних клітин у м’язах, які у відповідь на реплікацію вірусних нуклеїнових кислот продукують інтерферон І типу, що зумовлює гіперактивацію імунокомпетентних клітин і розвитку аутоімунних реакцій [28]. Сьогодні продовжує вивчатися роль природних кілерів [29] в патогенезі ЮДМ, а також PTPN22 (варіант R620W якого пов’язаний з підвищеним ризиком розвитку хвороби). Зазначені досягнення зумовили спроби використання з лікувальною метою блокаторів інтерферонів та молекул, які здатні блокувати гіперактивацію макрофагів та природних кілерів, у тому числі сіаловмісний імуноглобуліноподібний лектин-1 (Siglec-1) [30]. Cyril Gitiaux припускає, що підвищення рівня інтерферону у хворих на ЮДМ та ефективність відповідної терапії можуть опосередковано підтверджувати роль вірусних інфекцій як тригерів розвитку ЮДМ [31].
Неуточненою залишається роль В-лімфоцитів у патогенезі даної хвороби. Оскільки вони утворюються в кістковому мозку протягом усього життя людини і беруть участь у формуванні та підтримці адаптивного імунітету, дефект В-клітинної толерантності призводить до синтезу аутоантитіл, які індукують розвиток запалення і наступну деструкцію тканин. Окрім цього, порушення В-клітинної костимуляції Т-лімфоцитів відіграє фундаментальну роль у розвитку ранніх стадій аутоімунної відповіді [32].
Визначальну роль у формуванні як гуморальних, так і клітинних аутоімунних реакцій при ЮДМ має явище мікрохімеризму [33]. Оскільки у деяких хворих у стінках внутрішньом’язових судин виявлені відкладення імуноглобулінів, припущена наявність специфічних аутореактивних антитіл.
Подальші дослідження показали, що продукція міозитспецифічних аутоантитіл корелює з носійством певних HLA-антигенів, характерних для ЮДМ.
Існують численні дослідження, які дозволяють виявити роль певних аутоантитіл у розвитку ЮДМ (табл.1) [34].
Таблиця 1. Частота виявлення аутоантитіл у хворих на ЮДМ та дорослих з дерматоміозитом/поліміозитом [34]
| Аутоантитіло | Антигени-мішені | Клінічні асоціації | % у дітей | % у дорослих |
|---|---|---|---|---|
| Загалом | 65 | 80 | ||
| Антисинтетазні (анти-ARS, Jo1) | Цитоплазматичні транспортні РНК-синтетази | Антисинтетазний синдром | <3
у старшому віці |
20–40
до 90% при антисинтетазному синдромі |
| Анти-Mi-2 | Ядерні гелікази | Шкірні і помірні м’язові ураження | 5–10 | 10–50 |
| Анти-SRP | Сигналрозпізнавальні ділянки | Поліміозит, гіперферментемія, некротизуюча міопатія, тяжкий дебют | <1 | 2 |
| Анти-ARS | Амінокислоти тРНК | Тяжке шкірне ураження, швидка прогресія | 2 | 20–30 |
| Анти-SAE | Убіквітинподібні ензими | Шкірні прояви | <1 | 5 |
| Анти-TIF1-γ (p 155/140) | Транскрипційний внутрішній фактор | Фотосенсибілізація, шкірні прояви, ліподистрофії
Новоутворення |
18–30 | 7–21 |
| NXP2/анти-MJ (p 140) | Ядерні матриксні білки | Кальциноз | 15–20 | 1,6–17 |
| Анти-MDA5 (CADM-140) |
Асоційовані з геном меланоми РНК-гелікази | Прогресуюче ураження легень у дорослих | 7–38 | 1–33 |
| Анти-HMCGR | 3-гідрокси-3-метилглутарилкоензим A-редуктаза | Застосування статинів, тяжкий перебіг | 1 | 6–70 |
| Анти-Сn1A | Дефосфорилази, нуклеозид монофосфат | Виражена слабкість | 11–35 | 4–21 |
Так, антисинтетазні антитіла (анти-ARS), мішенню щодо яких є цитоплазматичні транспортні РНК-синтетази, відмічають рідше у дітей з ЮДМ, як і антитіла до ядерних геліказ (анти-Mi-2-аутоантитіла), які виявляють при виражених шкірних і помірних м’язових проявах. Відомо, що більш ніж у 25% дорослих із запальною міопатією виявлення TIF1-γ і анти-NXP-2 пов’язані з виникненням злоякісної пухлини протягом року [35]. У той же час анти-TIF1-γ (p 155/140) аутоантитіла, які у дорослих виявляють при паранеопластичних дерматоміозитах, відмічають в дитячому віці частіше та асоційовані з розвитком неоплазій значно пізніше дебюту ЮДМ, вже у дорослому віці [35]. У дітей японського походження частота виявлення антитіл до гена меланоми MDA-5 при ЮДМ сягає 28% [36–38] порівняно з 6% у країнах західного світу [39]. У дітей віком до 5 років аутоантитіла до ядерних матриксних білків (анти-MJ (p 140)-автоантитіла) часто асоційовані з кальцинозом та розвитком тяжких деструктивних уражень шлунково-кишкового тракту з кровотечами [40, 41].
Загалом виявлення аутоантитіл при ЮДМ відмічають рідше, проте воно асоційоване з більш тривалим і тяжким перебігом захворювання [42, 43]. Будь-які типи аутоантитіл виявляють при ЮДМ протягом усього періоду спостереження не в усіх хворих, що суттєво обмежує їх діагностичне значення. У пацієнтів з незвичайним перебігом хвороби рекомендується визначення також міозитасоційованих антитіл (анти-PmScl, анти-U1-RNP, анти-La (SSB), анти-Ro (SSA) та анти-Sm), що може бути корисним для уточнення діагнозу у випадках з перехресними синдромами. У 80% випадків у сироватці крові дітей з ЮДМ реєструють антинуклеарні антитіла [44], що може пояснювати їх схильність до розвитку полівалентних аутоімунних реакцій.
Зазначене призводить до вищої частоти виявлення неревматичних аутоімунних хвороб у пацієнтів з ЮДМ, які не завжди своєчасно виявляють. З одного боку, це може бути зумовлено тим, що їх симптоми маскуються призначеною при ЮДМ імуносупресивною та протизапальною терапією [45, 46], з іншого — множинні аутоімунні захворювання є характерним проявом численних синдромів порушення імунної регуляції (мутації в гені AIRE, гаплонедостатність CTLA-4 або LRBA та посилення функції Stat3 тощо) [47]. Проспективний аналіз когорти з 590 пацієнтів реєстру ЮДМ в дитячій лікарні імені Х. Лурі у Чикаго, США, продемонстрував, що частота множинного аутоімунітету без урахування overlap-синдромів з іншими ревматичними хворобами становила 8,7%, найчастішими серед решти виявлених у пацієнтів з ЮДМ аутоімунних захворювань були вітиліго, целіакія та тиреоїдит [46]. Узагальнені основні патогенетичні прояви ЮДМ представлені на рис. 2.
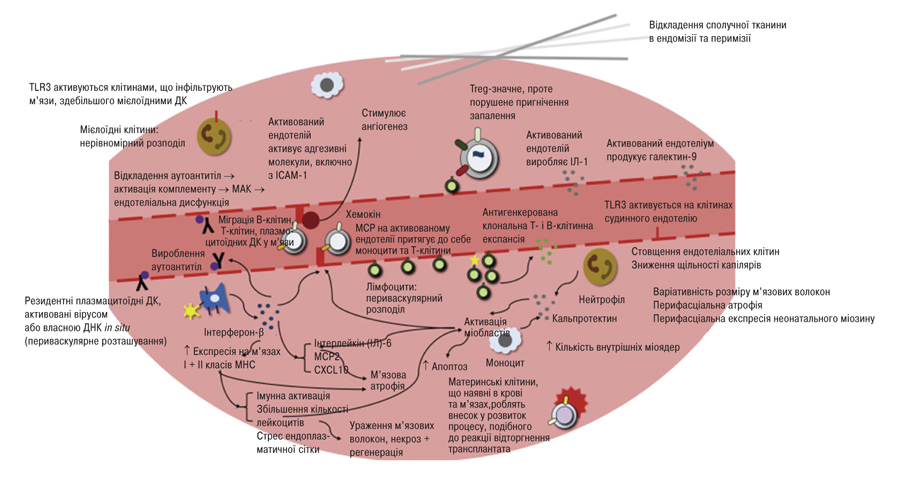
Патоморфологія
Гістологічне дослідження біоптатів мʼязів на перших етапах міозиту дозволяє виявити їх інфільтрацію лімфоцитами, ознаки васкуліту, вакуолізацію ендотелію, тромбози, ділянки некрозу мʼязових волокон, набряк, гіперплазію, посилення фагоцитозу зруйнованих міоцитів і регенерацію мʼязових фібрил. Фіброз та жирову інфільтрацію відмічають при проведенні дослідження в більш пізніх стадіях хвороби. Проте дослідження біоптатів шкіри, навіть в неуражених ділянках, також може характеризуватися васкулітом у різних стадіях його розвитку. Саме вираженість васкуліту відрізняє ЮДМ від дерматоміозиту у дорослих і дозволяє відокремити гострий процес (з наявністю інфільтрації ТCD45RA+-лімфоцитами, CXCL13 та зазначеними змінами судин) від хронічного ураження (коли відмічають зменшення кількості капілярів, явища фіброзу, м’язові інфаркти) [48–50]. Саме тому у випадках пізньої проблемної диференційної діагностики в дитячому віці зберігається сенс використання біопсії шкіри для уточнення діагнозу або тактики подальшого лікування.
Особливості клінічної картини ЮДМ
Клінічні ознаки і симптоми ЮДМ, як і всіх системних захворювань сполучної тканини, різноманітні. Досі діагностика хвороби ґрунтується здебільшого на результатах клінічного та лабораторного обстеження. За даними Patwardhan та співавторів діагноз ЮДМ може затримуватися майже на пів року (так, вік дебюту становить в середньому 81,97±46,63 міс, тоді як вік діагнозу 87,97±45,9 міс) [51]. Причому у дітей молодшої вікової групи тривалість затримки діагнозу ще більша (5,6 міс проти 4,5 міс відповідно) [52]. Безумовно, пізніше діагностуються більш легкі форми захворювання [53].
Клінічна картина ЮДМ може істотно варіювати залежно від варіанту його дебюту. Найчастішими скаргами в дебюті хвороби у дітей є біль у м’язах (18%); слабкість (82,1%); порушення ходи; порушення ковтання (10,3%); дистонія (14%); висип (82% — геліотропний, 56,4% — малярний), виникають реакції фоточутливості (7,7%), оральні виразки (2,6%), артралгії (18%) або артрит (12,8 %) [54]. Проте є деякі скарги, які не відмічають у дорослих хворих, наприклад, зростання частоти респіраторних захворювань. Також у дитячому віці частіше виявляють гострий початок захворювання з лихоманкою, вираженим больовим соматичним синдромом та зменшенням маси тіла [34, 54]. Лише в одиничних випадках, на відміну від дорослих, при ЮДМ в дебюті відмічають синдром Рейно.
Провідним у клінічній картині ЮДМ є міопатичний синдром. Він складається з міалгій, болючості та зміни консистенції м’язів при пальпації, м’язової слабкості [55, 56]. У подальшому у дітей швидко розвивається гіпотрофія м’язів.
М’язова слабкість зумовлена недостатністю скоротливої функції м’язів і є найчастішим першим проявом міопатичного синдрому у дітей. Вона характеризується значним обмеженням самостійних рухів: дитина не може самостійно встати, сісти, підняти ногу на сходинку, утримувати якийсь предмет у руці, розчісуватися, одягтися без допомоги тощо. Проблемою виявлення м’язової слабкості у дітей молодшого віку є відсутність можливості самостійно поскаржитися, часто батьки тривалий час неправильно трактують симптоми, вважають дитину лінивою, тихою (дитина сидить, не грає в активні ігри) або надмірно активною (внаслідок чого дитина падає). Поступово може розвинутися ураження м’язів шиї, при якому діти не можуть навіть утримувати голову, з приводу чого батьки звертаються до лікарів-неврологів. Залучення до процесу м’язів глотки призводить до дисфагії, коли виникає поперхування під час ковтання, затікання їжі в носову порожнину, аспірація. Ураження м’язів гортані викликає дисфонію з гугнявим відтінком голосу та охриплістю, яку також часто вважають проявом хронічної патології носоглотки. Найважчим при ураженні скелетних м’язів є ураження міжреберних м’язів та діафрагми, яке призводить до зменшення життєвої ємності легень і розвитку вторинних ускладнень з боку органів дихання запального характеру, внаслідок чого діти спостерігаються тривало пульмонологами або імунологами з приводу рецидивних пневмоній.
Клінічні ознаки, пов’язані з системними васкулопатіями, є центральними у діагностиці ЮДМ. При ЮДМ шкірний синдром у вигляді дерматиту на відкритих частинах тіла є досить частим симптомом. Еритема при ЮДМ найчастіше локалізується на обличчі між верхньою повікою та бровою, часто поєднана з набряком навколо очей. Такі шкірні зміни відрізняються значною стійкістю, синюшним відтінком, можуть супроводжуватися лущенням і свербежем [57]. Іноді дерматит має сквамозний характер і нагадує атопічний дерматит, себорею або псоріаз, через що пацієнти можуть тривало затримуватися під спостереженням дитячих алергологів та дерматологів. Висип при ЮДМ може поширюватися на шию і грудну клітку, верхню частину спини і верхні кінцівки, живіт. Дуже характерні еритема фіолетового кольору на розгинальних поверхнях верхніх кінцівок у вигляді широкої смуги, деревовидне ліведо, периферичні набряки кистей та стоп, почервоніння навколонігтьових валиків та симптом Готтрона. Висип у дітей може бути поліморфним: водночас на шкірі є різні елементи та вогнища гіпо- і гіперпігментації (пойкілодермія). Можливі вогнищева або тотальна алопеція та дистрофічне ураження нігтів. Встановлено, що початок висипу та міозиту може бути неодночасним при ЮДМ [58]. Останні роботи свідчать про можливість шкірного ураження у 17,9% в дебюті зі шкірними проявами без м’язової слабкості [3]. Вважається, що ізольоване чи передуюче шкірне ураження при ЮДМ більше притаманне особам азійської популяції. Прогностичні чинники прогресування аміопатичного ЮДМ досі чітко не ідентифіковані. Дослідниками відмічено, що у 42,86% випадків ізольованого шкірного ураження можуть протягом тривалого часу (більше 10 років спостереження) не розвиватися м’язове ураження чи підвищення активності ферментів міолізу в сироватці крові [59].
Патогномонічним для ЮДМ є таке специфічне ураження, як поява кальцинатів у м’язах. Як правило, кальцинати виявляють у ділянці нижніх кінцівок (литкові мʼязи, стегна), в місцях гострої або хронічної травматизації м’язів (на ліктях, колінах, пальцях), в місцях внутрішньом’язових ін’єкцій (на сідницях) [60]. Формування множинних кальцинатів додатково ускладнює рухи дитини та може супроводжуватися утворенням нориць, з приводу яких проводяться неодноразові хірургічні втручання.
Ураження суглобів у хворих на ЮДМ характеризується розвитком недеструктивних артритів, найчастіше дрібних суглобів, зі швидким формуванням сухожилково-м’язових контрактур [61].
Ураження внутрішніх органів при ЮДМ виявляють рідше, ніж при інших ревматичних хворобах.
Ураження легень може проявлятися у вигляді гострого дифузного альвеоліту, інтерстиціального легеневого фіброзу, експіраторної задишки при ураженні діафрагмальних м’язів, вторинних неспецифічних пневмоній. Ураження легень є основною причиною летальності при ЮДМ [62, 63].
Ураження серця можуть мати різний характер — від синусової тахікардії до тяжкого міокардиту з ураженням провідної системи серця, порушеннями ритму і застійною серцевою недостатністю [64].
Судинні порушення відзначаються практично у кожного хворого на ЮДМ, проте в різному ступені, представлені капіляритами долонь, сітчастим ліведо або інфарктами нігтьового ложа. Генералізоване ураження судин характерне для дітей дошкільного віку, часто супроводжується болем в уражених зонах, некрозами, виразковими і гнійними процесами [65].
Ураження нирок виявляють рідко, їх прояви коливаються від протеїнурії до нефротичного синдрому і гострої ниркової недостатності [66, 67].
Ураження нервової системи відмічають частіше, переважно у вигляді поліневритів, зрідка — енцефалітів із судомними синдромами і вогнищевою неврологічною симптоматикою [68].
Ураження органів травлення найчастіше проявляється порушенням моторики стравоходу з ознаками дисфагії, рідше (при системному васкуліті) розвиваються перфорація кишечнику, шлунково-кишкові кровотечі і жировий гепатоз [69].
Ураження слизових оболонок відмічають у всіх хворих на ЮДМ, найчастіше розвиваються специфічний гінгівіт, рідше — кон’юнктивіт, набряк піднебіння і голосових зв’язок, що призводить до дисфонії, можуть виникати вульвіти і риніти [70].
У разі високої активності ЮДМ відзначається лімфопроліферативний синдром.
Симптоматика і перебіг ЮДМ відрізняються від дерматоміозиту у дорослих пацієнтів, зокрема, залученням дистальних м’язів кінцівок, що характерно для дітей молодшого віку при тяжкому перебігу хвороби; більш високою частотою розвитку тяжкого кальцинозу; частим формуванням стійких сухожильно-м’язових контрактур; відсутністю асоціації з неопластичним процесом (табл. 2).
Табл. 2. Відмінності спектру клінічних проявів ЮДМ та дерматоміозиту у дорослих [67, 73, 74]
| Клінічний симптом | Частота виявлення клінічного симптому | |
|---|---|---|
| ЮДМ (%) | Дерматоміозит (за C.M. Pearson) (%) | |
| Лихоманка | 16–39,3 | |
| Ураження м’язів | 100 | 100 |
| Слабкість | 92–95 | 100 |
| Біль | 25 | 49 |
| Атрофії | 34,2 | 53 |
| Ураження суглобів | 23–64 (артрит 6) | 32 |
| Контрактури | 9 | 32 |
| Ураження шкіри | 70–94 | 40 |
| Ураження слизових оболонок | 6 | |
| Кальциноз | 30–40 (>ПМ, >африканського походження) | 10–20 |
| Ураження легень | 5–50
Інстерстиціальне ураження 3,8 |
80 |
| Ураження серцево-судинної системи | 32,8 за даними інструментальних досліджень (вірогідно рідше артеріальна гіпертензія) | 20–50 |
| Синдром Рейно | 9,1 | 21 |
| Васкуліт | 2,6 | |
| Гастроінтестинальні ураження | 5–37 | 49 |
Важливим для розуміння відмінності клінічних проявів при ЮДМ від дерматоміозиту у дорослих пацієнтів є частий розвиток окремих ускладень: затримки фізичного розвитку як за масою тіла, так і за лінійним ростом, більш частий розвиток інтерстиціальної хвороби легень; набряків, випадіння волосся, порушення серцевої провідності, зниження щільності кісткової тканини; ліподистрофії [59, 67, 71, 72].
Як і у дорослих пацієнтів, першим та стандартним кроком діагностики є дослідження активності м’язових ферментів у сироватці крові.
Оскільки розвиток ЮДМ у дітей молодшої вікової групи відбувається на тлі переважання неспецифічної імунної відповіді та під більшим впливом генетичних чинників, закономірно, що перебіг хвороби у дітей молодшого віку матиме особливості.
Ретроспективним дослідженням Sarah Tansleya and Lucy R. Wedderburnc [34] показано, що особливостями ЮДМ у дітей молодшого віку було переважання дівчаток, відсутність сезонності дебюту, більш частий лихоманковий синдром (73% проти 35% у дітей віком старше 3 років на момент дебюту), майже вдвічі вища частота родинного анамнезу, що обтяжений аутоімунною патологією, рідше виявлення геліотропної еритеми (не виявлялася у 36% у віці до 3 років проти 13% у дітей старшого віку), синдром Готтрона (відсутній у 47% дітей молодшого віку проти 6,8% у дітей віком старше 3 років на момент дебюту), зміни капіляроскопії (не виявлено у 63% дітей молодшої вікової групи), нижчою була активність креатинкінази (у 73,6% залишалась в межах норми), аспартатамінотрансферази (нормальні рівні відмічені у 76,3% дітей віком до 3 років) або альдолази (у 78,9% спостережені нормальні показники) [34]. Проте у дітей молодшого віку авторами відмічено частіший розвиток анемії (47%), підвищення швидкості осідання еритроцитів (ШОЕ) (63%) [34].
Визначення активності та оцінка поліпшення стану при ЮДМ
Сьогодні дитячі ревматологи залучені в численні дослідження щодо оптимізації кількісного визначення активності ревматичних хвороб. Педіатричною ревматологічною міжнародною дослідницькою організацією (Paediatric Rheumatology INternational Trials Organisation — PRINTO) запропоноване рутинне використання дитячими ревматологами 4 наступних критеріїв клінічно неактивного захворювання, з яких у дитини з ЮДМ мають бути наявні 3:
1. Активність креатинкінази в сироватці крові 150 МО/л,
2. Оцінка за дитячою шкалою оцінки міозиту (CMAS, яка за 14 позиціями кількісно за часовими проміжками чи якісно оцінює можливості дитини утримувати та піднімати кінцівки/голову, виконувати самостійно побутові рухи) ≥48 балів,
3. Ручне тестування м’язів 8 груп (ММТ-8) ≥78;
4. Оцінка лікарем стану хворого 0–2 [75].
CMAS та MMT-8 — основа більшості клінічних оцінок сили та витривалості м’язів у дітей з ЮДМ. Стандартизація CMAS для досягнення 52 балів була розрахована на здорових дітей віком 4–9 років [76]. Проте у здорових дітей віком 4–5 років зазвичай досягається лише 46 балів [77]. А оскільки у 26,3% випадків ЮДМ діагностується у віці до 4 років, інтерпретація даних CMAS може бути утруднена [78].
Для пацієнтів віком 5–7 років розроблена окрема система оцінки (PROMIS), причому встановлено, що оцінка стану дитини батьками не замінює повністю оцінку свого стану самою дитиною [79–81].
Проведене порівняльне дослідження показало, що критерії покращення, запропоновані Міжнародною групою з оцінки та клінічного вивчення міозиту (IMACS), більш чутливі при ЮДМ, ніж у дорослих хворих на дерматоміозит (99% проти 89% для мінімального покращання), причому у дітей меншу вагу має оцінка ММТ-8, більшу — оцінка CMAS, глобальна оцінка лікаря та активність ферментів міолізу в сироватці крові [82].
Сьогодні при ЮДМ пропонується визначати ряд додаткових імунологічних біомаркерів, включаючи інтерферон-α, інтерлейкін-17 і мутації у генах-регуляторах в якості потенційних маркерів активності ЮДМ [14, 83].
Крім відомих анти-Ro-52, у дітей з ЮДМ виявлені нові біомаркери, асоційовані з тяжким перебігом, розвитком інтерстиціального ураження легень та дигітальними виразками, зокрема такі, як інтерлейкін-6, -18 та феритин [38, 84–86].
Дитячим ревматологам варто співпрацювати з суміжними фахівцями з приводу психологічних проблем у дітей з ЮДМ (підвищується неспокій, розвиваються психотичні реакції, відмічаються десоціалізація, зниження рівня освіти та перспектив пацієнтів).
Класифікаційні критерії
У дитячому віці хронічні ідіопатичні запальні міопатії, до яких належить ЮДМ, є відносно неоднорідними порушеннями. Згідно з Міжнародною класифікацією хвороб (МКХ) Х перегляду ЮДМ віднесений до системних захворювань сполучної тканини (дерматополіміозит М.33 (М33.0 — ювенільний дерматоміозит)). Загальноприйнятим є виокремлення серед ЮДМ форм з ураженням тільки м’язів (поліміозит). Згідно з МКХ ХІ перегляду ЮДМ належатиме до рубрики 4A41 ідіопатична запальна міопатія:
4A41.01 — ювенільний дерматоміозит;
4A41.0Z — дерматоміозит неуточнений;
4A41.1 — поліміозит.
В якості критеріїв діагнозу ЮДМ досі використовуються критерії за Bohan and Peter, 1975, чутливість яких у дітей становить 98,9%, специфічність — 90,3% [87, 88].
Після того як Міжнародним проєктом критеріїв класифікації міозитів (International Myositis Classification Criteria Project — IMCCP)/Американським коледжем ревматології (American College of Rheumatology — ACR/Європейським альянсом асоціацій ревматологів (European Alliance of Associations for Rheumatology — EULAR) у 2016 р. запропоновані нові класифікаційні критерії запальних міопатій, у 2018 р. педіатрична ревматологічна міжнародна дослідницька організація PRINTO ініціювала розробку нових класифікаційних критеріїв ЮДМ, до яких запропоновано включити результати магнітно-резонансної томографії (МРТ) м’язів, оцінку кальцинозу, наявність виразок, опис біопсії, наявність дисфагії, дистонії, міалгії, деталізацію змін електроміографії (ЕМГ), виявлення міозитасоційованих аутоантитіл, дані ультразвукової діагностики (УЗД) м’язів, капіляроскопії, рівня неоптерину. Проте виявлено, що релевантними при ЮДМ залишаються лише патогномонічний висип на шкірі, підвищення ферментів міолізу та м’язова слабкість [89–92].
Диференційна діагностика ЮДМ відрізняється від диференційного пошуку у дорослих та часто починається з виключення зовнішніх, позам’язових причин м’язової слабкості — порушень електролітного обміну (зниження вмісту в крові калію, кальцію, магнію), ендокринних захворювань (гіпертиреоз, гіперпаратиреоз, гіпофосфатемія, порушення обміну вуглеводів та інших хронічних станів, що супроводжуються тканинною гіпоксією), синдрому мальабсорбції з дефіцитом вітамінів Е і D. Також у дітей молодшого віку слід обов’язково виключати незапальні міопатії (м’язові дистрофії; міопатії; міотонічні синдроми; періодичні паралічі; хвороби з переважним ураженням рухового нейрона) та хвороби, зумовлені порушенням метаболізму в м’язах, перш за все, порушенням утилізації глюкози або жирів (наприклад глікогеноз). Важливо пам’ятати, що гострий виражений больовий синдром з виключенням активних рухів може виникати у дитини з інфекційною полінейропатією. У разі виявлення ознак міолізу і запального процесу в м’язах диференційний діагноз проводиться з іншими міопатіями (при поліомієліті, токсоплазмозі, трихінельозі, цистицеркозі, ехінококозі, а також туберкульозних та вірусних міозитах тощо).
Підходи до терапії ЮДМ. До винайдення глюкокортикоїдів (ГК) третина дітей з ЮДМ помирали одразу, третина мали кальциноз, лише третина виживали, проте мали тяжкі наслідки [34]. Мала кількість хворих на ЮДМ і досі суттєво утруднює проведення клінічних досліджень та призводить до спроб спонтанного застосування лікарських засобів. З 1970-х років стандартним лікуванням ЮДМ було щоденне введення високих доз пероральних ГК, що продовжувалося до досягнення клініко-лабораторного поліпшення, далі вкрай повільно проводилося зниження добової дози. Така схема призначення ГК зберігає актуальність у дитячій ревматологічній практиці й досі. Причому загальний курс ГК має тривати не менше 2 років. Внаслідок такого тривалого призначення у більшості пацієнтів розвиваються побічні ефекти терапії ГК (асептичні некрози кісток, остеопороз та затримка росту тощо) [93–96].
В останні роки з’явилися перші протоколи лікування ЮДМ. Альянс досліджень дитячого артриту та інших ревматичних хвороб (CARRA) запропонував початкові рекомендації щодо лікування ЮДМ середньої тяжкості та терапію переважно шкірного захворювання [97].
За даними отриманого фактичного застосування ліків у 320 дітей з ЮДМ переглянуто лікування та створені ACR-рекомендації (2016 р.) та Консенсусні рекомендації EULAR з ведення хворих на ЮДМ (2017 р.), які уніфікували підходи до їх лікування. В останніх зазначено, що лікування пацієнтів з ЮДМ є складною клінічною задачею та повинно відбуватися в спеціалізованих багатопрофільних центрах (4D, 100%). Крім контрольованого фізичного навантаження та дієти, однією з важливих загальних рекомендацій є використання сонцезахисних засобів (4 D, 100%) [98].
Препаратами вибору медикаментозної терапії ЮДМ залишається призначення високих доз ГК (пероральне або внутрішньовенне введення) (1B-A, 100 %). ГК призначають при встановленні діагнозу ЮДМ усім хворим. Високі дози ГК (≥1 мг/кг маси тіла) слід призначати вже при середньо-тяжких формах ЮДМ (2A B, 100%). Слід надати перевагу внутрішньовенному їх введенню, якщо є сумніви щодо можливості їх абсорбції (3 С, 100%). Показаннями до проведення пульс-терапії при ЮДМ є збільшення вираженості дисфагії, розвиток альвеоліту, неможливість усунення міопатичного кризу стандартними дозами ГК. Залишається рекомендованим тривале призначення стартової дози, яке може становити 1–4 і більше місяців (4D, 100%). Категорично протипоказана повна відміна кортикостероїдів, які призначені у дозі 1 мг/кг/добу за преднізолоном і вище протягом 2–4 міс після досягнення терапевтичного ефекту. Також доцільно відмітити, що у дітей з ЮДМ може погіршуватися всмоктування перорального преднізолону, що корелює з показниками капіляроскопії [99]. Рекомендується застосовувати захист шлунка блокаторами H2-рецепторів та інгібіторами протонної помпи.
Доведено, що ГК при ЮДМ знижують смертність з 30 до 5% [94, 100]. Найбільш доцільно лікування ЮДМ в тяжких випадках починати з внутрішньовенного введення високих доз метилпреднізолону, що задокументовано в багатонаціональному дослідженні PRINTO [101]. Побічні реакції пульс-терапії у дітей можуть бути специфічними і не бути ідентичними небажаним явищам у дорослих пацієнтів (гінекомастія, виражені трофічні порушення шкіри, психотичні реакції з вираженою агресією, інфекційні захворювання, що викликані атиповою флорою) [102, 103].
Показаннями до призначення засобів базисної хворобомодифікуючої терапії у дітей з ЮДМ є наявність маркерів несприятливого прогнозу, недостатня ефективність або неможливість підвищення дози ГК. Ці рекомендації свідчать, що додавання метотрексату або циклоспорину А до терапії ГК призводить до кращого контролю захворювання, ніж монотерапія ГК, а кращий профіль безпеки сприяє вибору комбінації метотрексату і преднізолону (1B, A, 100%) [98]. Введення метотрексату після 1997 р. дозволило використовувати нижчі дози ГК для контролю захворювання [104, 105]. Останніми рекомендаціями підкреслюється, що застосування метотрексату слід починати з дози 15–20 мг/м2 на тиждень (максимальна доза не має перевищувати 40 мг на тиждень). Варто зауважити, що при даній патології дітям протипоказане внутрішньом’язове введення метотрексату, бажано в дебюті хвороби призначати його підшкірно (4 D, 100%) [98]. При дотриманні техніки введення кальцинати в місцях введення не формуються, проте ефективність препарату і темпи розвитку ефекту істотно вищі порівняно з пероральним прийомом метотрексату через високу частоту ураження органів травлення при ЮДМ, що зумовлює зниження біодоступності препаратів, які застосовуються перорально.
Пацієнти з факторами високого ризику повинні проходити обстеження на наявність гепатиту В і С, при виявленні яких доцільно замінити метотрексат на інший препарат базисної терапії [106]. Також до несприятливих ефектів метотрексату належать ураження респіраторного тракту та зупинка росту у дітей [107].
Якщо у пацієнта відмічено неадекватну відповідь на терапію, інтенсифікація лікування з включенням хворобомодифікуючих протиревматичних препаратів повинна проводитися протягом перших 12 тиж від дебюту хвороби після консультації з експертним центром (4 D, 100 %) [98].
У пацієнтів, які не переносять метотрексат, його замінюють на інший базисний препарат (циклоспорин А або мікофенолату мофетил) (3С, 100%). Mікофенолату мофетил може бути корисною терапією при ураженнях м’язів та шкіри (включаючи кальциноз) (3С, 100%) [98].
Мікофенолату мофетил рекомендують призначати кожні 12 год в дозі 20 мг/кг маси тіла (максимальна добова доза не має перевищувати 1000 мг), проте не існує точних даних, в кого з хворих на ЮДМ він матиме переваги [108, 109]. Хоча це не є загальновизнаним, призначення препаратів вітаміну D може знизити ефективність мікофенолату мофетилу при ЮДМ [110], що є приводом для занепокоєння, оскільки підтримка рівня вітаміну D в організмі хворих на ЮДМ є критично важливою.
Такролімус значно рідше застосовували при ЮДМ, відмічено, що він був ефективним у пацієнтів з антитілами до HMGCR [111, 112].
Циклоспорин А є засобом другої лінії при ЮДМ і призначається лише тоді, коли інші препарати не дали результатів, особливо при стійкому шкірному синдромі [113]. Нещодавнє рандомізоване клінічне дослідження 139 пацієнтів з ЮДМ у 22 країнах порівняло 3 плани лікування: після пульс-терапії метилпреднізолоном діти отримували або 2 роки перорального застосування преднізолону, або преднізолон з метотрексатом, або комбінацію преднізолону з циклоспорином А. Аналіз отриманих результатів показав, що комбінована терапія в обох випадках виявилася кращою порівняно з монотерапією преднізолоном, але більша кількість побічних реакцій відмічена при застосуванні циклоспорину А [114]. Також дослідники відмітили, що при підвищеному рівні ліпідів у сироватці крові збільшувався кліренс циклоспорину А, що зумовлювало зниження його ефективності [114].
Постійні прояви з боку шкіри та кальциноз відображають активність захворювання, тому їх також слід усувати за рахунок підвищення системної імунодепресії (4D, 3С, 100%) [98].
Для пацієнтів з тяжким ступенем активності захворювання (таким як тяжкі залучення внутрішніх органів або виразкове ураження шкіри) слід розглянути можливість додавання до лікування внутрішньовенного циклофосфаміду (3С, 100 %). Цей препарат успішно застосовується для лікування тяжких ЮДМ у дозі 500 мг/м2 кожні 2 тиж 3 рази, потім призначається 750 мг/м2 кожні 3–4 тиж шляхом 3–4 введень [115]. На жаль, циклофосфамід демонструє вкрай руйнівний вплив на репродуктивну систему та кардіотоксичний ефект [116, 117], що небезпечно й обмежує його застосування у дитячому віці.
Гідроксихлорохін при ЮДМ самостійно не викликає достатнього ефекту [118], проте його вплив на імунну систему (зниження синтезу інтерлейкінів-1, -6 та фактора некрозу пухлин-α, здатність блокувати TLR7/9 на дендритних клітинах плазмоцитів, зменшуючи викид інтерферону-1), антитромботичний ефект, нормалізація ліпідного обміну зумовлюють продовження його застосування при ЮДМ на етапі звуження терапії [118–120].
Препарати внутрішньовенних імуноглобулінів можуть бути корисними у випадках резистентного захворювання, особливо коли провідним є ураження шкіри (2В-4 С, 100%) [98]. Передбачуваний механізм їх дії полягає в осадженні мембрано-атакуючого комплексу C5beC9 на ендомізії капілярів. Внутрішньовенні імуноглобуліни рекомендується вводити по 1–2 г/кг маси тіла щомісяця [121], сьогодні в світі використовують форми для підшкірного введення для можливості підвищення доз [122], які в Україні ще не доступні. Рутинне їх застосування та призначення при інтерстиціальному ураженні легень не рекомендується [123]. Після його застосування у дітей шкірні прояви можуть зберігатися [97].
Враховуючи вищеописані імунологічні особливості у хворих на ЮДМ, виникла ідея щодо доцільності застосування біологічної терапії, зокрема, препаратів, мішенню для яких є В-лімфоцити, що можуть потенційно модулювати синтез патогенних аутоантитіл. Ще у 2002 р. вперше Levinе опублікував інформацію про застосування ритуксимабу при лікуванні дерматоміозиту. Подальші описи його призначення сприяли тому, що у 2012 р. він був рекомендований ACR для застосування при ЮДМ у випадках, які були рефрактерні до терапії ГК, цитостатичними засобами і внутрішньовенними імуноглобулінами. У 2013 р. С.V. Oddis навів результати оцінки ефективності і безпеки ритуксимабу за результатами рандомізованого подвійного сліпого плацебо-контрольованого дослідження у дорослих і дітей з активним міозитом, а вже у 2016 р. опубліковані нові консенсусні рекомендації з ведення хворих на ЮДМ, в яких запропоновано розглядати ритуксимаб як ад’ювантну терапію для пацієнтів з рефрактерним до попередньої терапії перебігом хвороби. Підкреслено, що досягнення результатів після його введення доцільно очікувати до 26 тиж (рівень доказовості 1B та D відповідно) [98, 124].
При застосуванні ритуксимабу у дітей рекомендований ретельний моніторинг сироваткового рівня імуноглобуліну G, особливо в перший рік (гіпогамаглобулінемія відмічена в 30–50% випадків), у деяких пацієнтів використовується замісна терапія внутрішньовенними імуноглобулінами [125]. Сьогодні ритуксимаб успішно застосовується для лікування пацієнтів із ЮДМ [126], його ефективність можна оцінити за оцінкою виявлення в В-клітинах кісткового мозку кругових ДНК (KREC) [127]. За першими звітами великого мультицентрового рандомізованого клінічного дослідження з оцінки ефективності ритуксимабу як у дорослих, так і у дітей із рефрактерними міозитами, виявлення у хворих міозитспецифічних антитіл (проти Jo-1 або Mi-2) та молодий вік пацієнтів (ЮДМ) передбачали кращу реакцію на ритуксимаб [127, 128].
Існують пропозиції щодо використання анти-ФНП-препаратів при ЮДМ. ФНП-α визнаний важливим цитокіном в патогенезі ЮДМ, його алель 308А асоціюється з більш стійким до імуносупресивної терапії перебігом захворювання та кальцинозом [129–131]. Анти-ФНП-терапія може бути призначена при тяжкому перебігу ЮДМ, причому доведено, що адалімумаб та інфліксимаб мають перевагу над етанерцептом (3D, 92%) [98].
У педіатричній практиці блокатори кальцієвих каналів застосовують при ЮДМ у разі наявності вісцеральної патології, синдрому Рейно, при легеневій та нирковій гіпертензії (або для їх профілактики). Зазвичай вазодилататори застосовують у поєднанні з дезагрегантами. В якості лікування кальцинозу найбільшу доказову базу сьогодні має застосування інгібіторів остеобластів [132].
У терапії ЮДМ широко використовують засоби симптоматичної терапії (засоби для лікування остеопорозу, репарантні, кардіотрофічні та гепатопротекторні препарати, вітамінотерапія, препарати гормону росту тощо) та хірургічне лікування. Свербіж можна лікувати місцево і перорально за допомогою антигістамінних препаратів.
Немає високого рівня доказів щодо часу припинення терапії ЮДМ; однак якщо пацієнту відмінені ГК і він знаходиться в ремісії хвороби, отримуючи метотрексат (або альтернативний базисний препарат) протягом мінімум 12 міс, може розглядатися питання припинення терапії (4D, 100%) [34, 98].
Напрямки подальших досліджень ефективності лікування
Зараз продовжуються дослідження щодо визначення ефективності та безпеки засобів біологічної терапії при ЮДМ.
Доведено, що абатацепт здатен зменшувати кількість активованих Т-лімфоцитів шляхом блокування сигналу через CD80 і CD85 від антигенпрезентаційної клітини, продовжується набір пацієнтів у дослідження його застосування при ЮДМ [133].
Спроби використання інгібіторів янус-кіназ базуються на їх властивостях пригнічувати синтез інтерферонів 1-го типу. Тофацитиніб (блокатор янус-кіназ 1-го і 3-го типу), барицитиніб та руксолітиніб (які блокують янус-кінази 1-го та 2-го типу), в даний час тестуються на терапевтичну ефективність при тяжкому дерматоміозиті у дорослих. Наразі є повідомлення, що барицитиніб успішно застосований при лікуванні кількох дітей із ЮДМ [134].
Ленабасум є пероральним синтетичним агоністом рецептора канабіноїдів 2-го типу, його використали в дослідженнях 2-ї фази для зменшення вираженості шкірних проявів [135].
Робоча група з питань ЮДМ в межах PReSS e-congress (вересень 2021 р.) проаналізувала дані 46 досліджень ЮДМ та дійшла висновку про необхідність валідаційних досліджень щодо чутливості біомаркерів ЮДМ, створення алгоритмів ескалації терапії, протоколів лікування тяжкого ЮДМ. Також запропоновано перегляд SHARE-гайдлайну з використанням стандартних процедур та огляд тяжких клінічних випадків.
Перебіг
Враховуючи нинішні можливості діагностики та лікування, прогноз ЮДМ суттєво покращився.
Перебіг ЮДМ демонстрував позитивну тенденцію за останні 60 років. Якщо до 1950-х років помирали третина пацієнтів [5, 87], то тепер смертність хворих на ЮДМ значно знизилася [7]. Прогностичних факторів все ще бракує, оскільки дизайн досліджень суттєво відрізняється та незіставний у різних дослідницьких центрах. Доведено, що стать і раса не є значущими для виживаності пацієнтів [34, 136].
Але, на жаль, прогноз у хворих на ЮДМ є не завжди сприятливим як щодо життя, так і формування тяжких ускладнень хвороби. Дослідниками відмічено, що у 34% пацієнтів зберігаються слабкість, у 53% — шкірні прояви, у 10–70% розвивається кальциноз (ризик якого зростає у хлопчиків віком до 5 років, в яких є aнти-NXP2-антитіла та зберігається ферментемія довше 6 міс), у 10–30% дітей з ЮДМ реєструються ліподистрофії (ризик яких вищий у разі виявлення порушень ліпідного обміну), 10% хворих на ЮДМ мають затримку фізичного розвитку [34].
При тривалому спостереженні пацієнтів A. Ravelli (2010) постійно активне захворювання зафіксовано у 41,2% пацієнтів, у 40,7% — знижену функціональну здатність, кальциноз — у 23,6%, ліподистрофії — у 9,7%.
Летальність при ЮДМ висока і становить 3,1%, тоді як у хворих з гострим ураженням легень сягає 74% [5]. Відмічено, що пацієнти молодшого віку на момент дебюту мали кращий прогноз, і через 5 років після встановлення діагнозу активне захворювання в них відмічали рідше (9% у дітей з дебютом у віці до 3 років порівняно з 35,7% у дітей, які захворіли у віці старше 3 років), і через 10 років (9% проти 45,1% відповідно) у них рідше виявляли тяжкі ускладнення, такі як остеонекроз [5].
Інші автори [54], які спостерігали 39 хворих на ЮДМ протягом більше ніж 10 років, відмітили, що у 70,97% дітей з ЮДМ після лікування отримано позитивну клінічну відповідь, але лише 19,4% досягли повної клінічної ремісії, у 33,33% пацієнтів відмічено моноциклічний перебіг та відсутність симптомів наприкінці спостереження, тоді як у решти виявлено ускладнення, а у 35% — рецидиви хвороби. Багатоваріантний аналіз показав, що жіноча стать, негативний знак Гауерса на початку захворювання та фоточутливість були сприятливими факторами досягнення повної клінічної ремісії.
Важливою сьогодні вважається розробка критеріїв оцінки індивідуального прогнозу пацієнта. За рекомендаціями Міжнародної асоціації з вивчення міозиту, до пацієнтів з високим ризиком несприятливого перебігу ЮДМ віднесені такі ознаки: тяжка функціональна недостатність (визначається при нездатності дитини самостійно встати з ліжка); низькі показники м’язової сили; наявність аспірації або дисфагії (до неможливості ковтати); шлунково-кишковий васкуліт (який виявлено інструментальними дослідженнями або на підставі наявності кров’яних випорожнень); міокардит; паренхіматозне ураження легень; ураження центральної нервової системи (зниження рівня свідомості або судоми); виразки шкіри; необхідність проведення інтенсивної терапії; вік дитини менше 1 року [4, 136–138].
На думку експертів, аналіз імунологічних особливостей у дитини також може дати змогу прогнозувати розвиток тих чи інших клінічних проявів хвороби та її перебіг. Проте доцільно відзначити, що сьогодні немає достатніх доказів для рекомендацій щодо визначення аутоантитіл для стратифікації ризику через відсутність перевірки даних у пацієнтів різних етнічних груп.
Узагальнюючи вищеописане, хотілося б відмітити, що ЮДМ розвивається у незрілому організмі дитини та має певні генетичні та імунологічні особливості, а також певні клінічні відмінності від дерматоміозиту дорослих: частіше — гострий або підгострий початок з лихоманкою, зменшенням маси тіла в дебюті захворювання, рідше відмічають перебіг з виключно м’язовим ураженням, більш поширені васкуліт, висока частота ульцерації, виражені міалгії, можливе залучення дистальних м’язів, більш часте залучення в процес внутрішніх органів, висока частота розвитку кальцинозу, формування стійких сухожильно-м’язових контрактур, відсутність асоціації з неопластичним процесом. Важливим є суттєве покращання прогнозу перебігу ЮДМ в останні роки, що призводитиме до збільшення частки хворих на дерматоміозит з початком у дитинстві серед усіх запальних міопатій.
Це обґрунтовує потребу у подальших дослідженнях менеджменту хворих із ЮДМ для покращення рівня надання медичної допомоги та вивчення віддалених наслідків хвороби.
Список використаної літератури
- 1. Cassidy J.T., Petty R.E., Laxer R.L. et al. (2010) From textbook of Pediatric Rheumatology E-Book: Expert Consult. 6th edition. New York: Elsevier Health Sciences.
- 2. Pilkington C. (2016) Handbook of Systemic Autoimmune Diseases. Chapter 10. Juvenile Dermatomyositis. Volume 11: 219–233. doi.org/10.1016/B978–0-444–63596–9.00010–4
- 3. Pachman L.M., Nolan B.E., DeRanieri D. et al. (2021) Dermatomyositis. New Clues to Diagnosis and Therapy. Curr. Treat Options in Rheum.; 7: 39–62. doi. 10.1007/s40674–020–00168–5
- 4. Ponyi A., Constantin T., Balogh Z. et al. (2005) Disease course, frequency of relapses and survival of 73 patients with juvenile or adult dermatomyositis. Clin. Exp. Rheumatol., 23(1): 50–56.
- 5. Ravelli, Trail L., Ferrari C. et al. (2010) Long-term outcome and prognostic factors if juvenile dermatomyostis. A multinational, multicenter study of 490 patients. Arthritis Care Res., 62: 63–72.
- 6. McCann L.J., Juggins A.D., Maillard S.M. et al. (2006) Juvenile Dermatomyositis Research Group:The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)-clinical characteristics of children recruited within the first 5 yr. Rheumatology (Oxford), 45(10): 1255–1260.
- 7. Mendez E.P., Lipton R., Ramsey-Goldman R. et al. (2003) NIAMS Juvenile DM Registry Physician Referral Group: US incidence of juvenile dermatomyositis, (1995–1998): results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum., 49(3): 300–305.
- 8. Huber A.M., Lang B., Leblanc C.M. et al. (2000) Medium and long-term functional outcomes in a multicenter cohort with juvenile dermatomyositis. Arthritis Rheum., 43: 541–549.
- 9. Ramanan A.V., Feldman B.M. (2002) Clinical outcomes in juvenile dermatomyositis. Curr. Opin. Rheumatol., 14(6): 658–662.
- 10. Pachman L.M. (2002) Juvenile dermatomyositis. immunogenetics, pathophysiology, and disease expression. Rheum. Dis. Clin. North A. 28(3): 579–602.
- 11. Reed A.M., Ytterberg S.R. (2002) Genetic and environmental risk factors for idiopathic inflammatory myopathies. Rheum. Dis. Clin. North Am., 28(4): 891–916.
- 12. Manlhiot C., Liang L., Tran D. et al. (2008) Assessment of an infectious disease history preceding juvenile dermatomyositis symptom onset. Rheumatology (Oxford), 47(4): 526–529.
- 13. Tezak Z., Hoffman E.P., Lutz J.L. et al. (2002) Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J. Immunol., 168(8): 4154–4163.
- 14. Wedderburn L.R., Rider L.G. (2009) Juvenile dermatomyositis. New developments in pathogenesis, assessment, and treatment. Best Pract. Res. Clin. Rheumatol., 23(5): 665–678.
- 15. Chen Y.W., Shi R., Geraci N. et al. (2008) Duration of chronic inflammation alters gene expression in muscle from untreated girls with juvenile dermatomyositis.BMC Immunol.; 9: 43.
- 16. Parks C.G., Wilkerson J., Rose K.M. et al. (2019) Association of ultraviolet 56 Pediatric Rheumatology (G Martini, Section Editor) myositis patient registry. Arthritis Care Res (Hoboken); epublished 1–5. An uptodate summary of an evolving field.
- 17. Neely J., Long C.S., Sturrock H. et al. (2019) Association of short-term ultraviolet radiation exposure and disease severity in juvenile dermatomyositis: results from the Childhood Arthritis and Rheumatology Research Alliance Legacy Registry. Arthritis Care Res. (Hoboken); 71(12): 1600–5.
- 18. Vidotto J.P., Pereira L.A., Braga A.L. et al. (2021) Atmospheric pollution: influence on hospital admissions in paediatric rheumatic diseases. Lupus. 21(5): 526–33.
- 19. Zhao C.N., Xu Z., Wu G.C. et al. (2019) Emerging role of air pollution in autoimmune diseases. Autoimmun Rev., 18(6): 607–14.
- 20. Mamyrova G., Rider L.G., Haagenson L. et al. (2005) Parvovirus B19 and onset of juvenile dermatomyositis. JAMA, 294(17): 2170–1.
- 21. Pachman L.M., Lipton R., Ramsey-Goldman R. et al. (2005) History of infection before the onset of juvenile dermatomyositis: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Research Registry. Arthritis Rheum(Arthritis Care & Research), 53(2): 166–72.
- 22. Vegosen L., Weinberg C., O’Hanlon T.P. et al. (2007) Seasonal birth patterns of myositis suggest a role for early enviornmental exposures in etiology. Arthritis Rheum., 56: 2719–28.
- 23. Rothwell S., Chinoy H., Lamb J.A. (2019) Genetics of idiopathic inflammatory myopathies: insights into disease pathogenesis. Curr. Opin. Rheumatol., 31(6): 611–6. An excellent review of the specific HLA alleles associated with inflammatory myopathy.
- 24. Rothwell S., Cooper R.G., Lundberg I.E. et al. (2015) Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann. Rheum. Dis.,10.1136(20819): 1–15.
- 25. Miller F.W., Cooper R.G., Vencovsky J.et al. (2013) Genome-wide association study of dermatomyositis reveals genetic overlap with other autoimmune disorders. Arthritis Rheum., 65(12): 3239–47.
- 26. Lintner K.E., Patwardhan A., Rider L.G. et al. (2016) Gene copy-number variations (CNVs) of complement C4 and C4A deficiency in genetic risk and pathogenesis of juvenile dermatomyositis. Ann. Rheum. Dis., 75(9): 1599–606.
- 27. Wilkinson M.G.L., Radziszewska A., Wincup C. et al. (2020) Using peripheral blood immune signatures to stratify patients with adult and juvenile inflammatory myopathies. Rheumatology (Oxford), 59(1): 194–204.
- 28. Piper C.J.M., Wilkinson M.G.L., Deakin C.T. et al. (2018) CD19(+)CD24(hi)CD38(hi) B cells are expanded in juvenile dermatomyositis and exhibit a proinflammatory phenotype after activation through tolllike receptor 7 and interferon-alpha. Frontiers in immunology [Internet], [PMC6024011]; 9: 1–15.
- 29. Throm A.A., Alinger J.B., Pingel J.T. et al. (2018) Dysregulated NK cell PLCγ2 signaling and activity in juvenile dermatomyositis. JCI insight, 3(22).
- 30. Lerkvaleekul B., Veldkamp S.R., van der Wal M.M. et al. (2021) Siglec-1 expression on monocytes is associated with the interferon signature in juvenile dermatomyositis and can predict treatment response Rheumatology (Oxford). pubmed.ncbi.nlm.nih.gov/?term=%22Rheumatology+%28Oxford%29%22%5Bjour%5Dhttps://www.ncbi.nlm.nih.gov/nlmcatalog?term=%22Rheumatology+%28Oxford%29%22%5BTitle+Abbreviation%5D. pubmed.ncbi.nlm.nih.gov/34387304/ (2021) Aug. 13; keab601. doi: 10.1093/rheumatology/keab601.Online ahead of print pubmed.ncbi.nlm.nih.gov/?term=%22Arthritis+Rheumatol%22%5Bjour%5D. http://www.ncbi.nlm.nih.gov/nlmcatalog?term=%22Arthritis+Rheumatol%22%5BTitle+Abbreviation%5D. pubmed.ncbi.nlm.nih.gov/28941175/
- 31. Gitiaux C., Latroche C., Weiss-Gayet M. et al. (2018) Myogenic Progenitor Cells Exhibit Type I Interferon-Driven Proangiogenic Properties and Molecular Signature During Juvenile Dermatomyositis Arthritis Rheumatol., Jan;70(1):134–145. doi: 10.1002/art.40328. Epub 2017 Nov 28. doi. 10.1002/art.40328
- 32. Vercoulen Y., Bellutti Enders F., Meerding J. et al. (2014) Increased presence of FOXP3+ regulatory T cells in inflamed muscle of patients with active juvenile dermatomyositis compared to peripheral blood PLoS One (2014). pubmed.ncbi.nlm.nih.gov/?term=%22PLoS+One%22%5Bjour%5Dhttps://www.ncbi.nlm.nih.gov/nlmcatalog?term=%22PLoS+One%22%5BTitle+Abbreviation%5D. pubmed.ncbi.nlm.nih.gov/25157414/. Aug 26;9(8):e105353. doi: 10.1371/journal.pone.0105353. eCollection
- 33. Ye Yi, van Zyl B., Varsani H. et al. (2012) on behalf of the Juvenile Dermatomyositis Research Group† Maternal microchimerism in muscle biopsies from children with juvenile dermatomyositis Rheumatology (Oxford). Jun; 51(6): 987–991.
- 34. Tansleya S., Wedderburnc L.R. (2017) Comparing and contrasting clinical and serological features of juvenile and adult-onset myositis: implications for pathogenesis and outcomes. Curr. Opin. Rheumatol., 27: 000–000, Ann Rheum Dis.; 76 (12): 1955–1964.
- 35. Fiorentino D.F., Chung L.S., Christopher-Stine L. et al. (2013) Most patients with cancerassociated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1gamma. Arthritis Rheum.; 65(11): 2954–62.
- 36. Tansley S.L., Simou S., Shaddick G. et al. (2017) Autoantibodies in juvenile-onset myositis: their diagnostic value and associated clinical phenotype in a large UK cohort. J. Autoimmun., 10(1016).
- 37. Kobayashi I., Akioka S., Kobayashi N. et al. (2020) Clinical practice guidance for juvenile dermatomyositis (JDM) (2018)-Update. Mod. Rheumatol.; 30(3): 411–23.
- 38. So H., Ip R.W., Wong V.T. et al. (2018) Analysis of antimelanoma differentiation-associated gene 5 antibody in Hong Kong Chinese patients with idiopathic inflammatory myopathies: diagnostic utility and clinical correlations. Int. J. Rheum. Dis., 21(5): 1076–81.
- 39. Tansley S.L., Betteridge Z.E., McHugh N.J. (2013) The diagnostic utility of autoantibodies in adult and juvenile myositis. Curr. Opin. Rheumatol., 25(6): 772–7.
- 40. Tansley S.L., Betteridge Z.E., Shaddick G. et al. (2014) Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (Oxford), 53(12): 2204–8.
- 41. Khojah A.M.L.V., Savani S.I., Morgan G.A. et al. (2020) Studies of 96 children with juvenile dermatomyositis: myositis specific antibody P155/140 (TIF-gamma) is associated with loss of nailfold capillaries, but not acquired lipodystrophy, determined by DXA. Arthritis Care Res (Hoboken). doi. org/10.1062/24535
- 42. Rider L.G., Shah M., Mamyrova G. et al. (2013) The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore); 92(4): 223–43.
- 43. Pachman L.M., Khojah A.M. (2018) Advances in juvenile dermatomyositis: myositis specific antibodies aid in understanding disease heterogeneity. J. Pediatr., 195: 16–27.
- 44. Reed A.M. (2021) Juvenile Dermatomyositis Workup Updated: May 12. emedicine.medscape.com/article/1417215-workup.
- 45. Prahalad S., McCracken C.E., Ponder L.A. et al. (2016) Familial autoimmunity in the Childhood Arthritis and Rheumatology Research Alliance registry. Pediatr Rheumatol Online J., 14(1): 14.
- 46. Malagón C., Gomez M.D.P., Mosquera C. et al. (2019) Juvenile polyautoimmunity in a rheumatology setting. Autoimmun Rev., 18(4): 369–81.
- 47. Gámez-Díaz L., Seidel M.G. (2021) Different Apples, Same Tree: Visualizing Current Biological and Clinical Insights into CTLA-4 Insufficiency and LRBA and DEF6 Deficiencies Front. Pediatr., 28 April. doi.org/10.3389/fped.2021.662645.
- 48. Duvvuri B., Pachman L.M., Morgan G. et al. (2020) Neutrophil extracellular traps in tissue and periphery in juvenile dermatomyositis. Arthritis Rheumatol., 72(2): 348–58.
- 49. Wienke J., Deakin C.T., Wedderburn L.R. et al. (2018) Systemic and tissue inflammation in juvenile dermatomyositis: from pathogenesis to the quest for monitoring tools. Front. Immunol., 9: 2951.
- 50. Papadopoulou C., McCann L.J. (2018) The vasculopathy of juvenile dermatomyositis. Front. Pediatr., 6: 284.
- 51. Patwardhan et al. (2012) Is juvenile dermatomyositis a different disease in children up to three years of age at onset than in children above three years at onset? A retrospective review of 23 years of a single center’s experience. Pediatric Rheumatology, 10: 34. http://www.ped-rheum.com/content/10/1/34.
- 52. McCann L.J. (2006) Тhe Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)–clinical characteristics of children recruited within the first 5 yr. [Електронний ресурс]. McCann L.J., Juggins A.D., Maillard S.M. et al (2006). Juvenile Dermatomyositis Research Group Rheumatology (Oxford), Vol. 45 (10: 1255.
- 53. Pachman L.M., Abbott K., Sinacore J.M. et al. (2006) Duration of illness is an important variable for untreated children with juvenile dermatomyositis. J. pediatr., 148(2): 247–53.
- 54. Sun C., Lee J.-H., Yang Y.-H. et al. (2015) Juvenile Dermatomyositis: A 20-year Retrospective Analysis of Treatment and Clinical Outcomes Chiang Pediatrics and Neonatology, 56: 31–39.
- 55. Dugan E.M., Huber A.M., Miller F.W. (2009) Photoessay of the cutaneous manifestations of the idiopathic inflammatory myopathies. Dermatol. Online J.,15(2): 1.
- 56. Проблемні питання дитячої ревматології (2019) Монографія за редакцією Антипкіна Ю.Г., Охотнікової О.М., Ошлянської О.А., Омельченко Л.І. 700 с.
- 57. Cao H., Parikh T.N., Zheng J. (2009) Amyopathic dermatomyositis or dermatomyositis-like skin disease: retrospective review of 16 cases with amyopathic dermatomyositis. Clin. Rheumatol., 28: 979–84.
- 58. Caproni M., Cardinali C., Parodi A. et al. (2002) Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch. Dermatol., 138: 23–7.
- 59. Arnson Y., Dovrish Z., Hadari R. et al. (2007) Amyopathic dermatomyositisdan uncommon presentation of dermatomyositis. Isr. Med. Assoc. J., 9: 492–3.
- 60. Wang A., Morgan G.A., Paller A.S. et al. (2020) Skin disease ismore recalcitrant thanmuscle disease: a longterm prospective study of 184 children with juvenile dermatomyositis. JAAD, in press.
- 61. Tse S., Lubelsky S., Gordon M. et al. (2001) The arthritis of inflammatory childhood myositis syndromes. J. Rheumatol. Jan; 28(1): 192–7. PMID: 11196524.
- 62. Prestridge A., Morgan G., Ferguson L. et al. (2013) Pulmonary function tests in idiopathic inflammatory myopathy: association with clinical parameters in children. Arthritis Care Res. (Hoboken); 65(9): 1424–31.
- 63. Mira-Avendano I.C., Parambil J.G., Yadav R. et al. (2013) A retrospective review of clinical features and treatment outcomes in steroid-resistant interstitial lung disease from polymyositis/dermatomyositis. [Електронний ресурс] SO Respir Med., Vol. 107 (6): 890–6.
- 64. Ghosh R., Roy D., Dubey S. et al. (2020) Juvenile dermatomyositis presenting as complete heart block in a 10-year-old girl. Paediatrics and international child health: 1–4.
- 65. Gorouhi F., Kiuru M., Silverstein M. et al. (2019) The pulseless patient: Profound vasculopathy as the presenting feature of fulminant dermatomyositis and response to therapy Jaad Case Report: Volume 5, Issue 2: 176–179.
- 66. Mohsin R.M., Tripath S.K., Phulware R.H. et al. (2019) Juvenile dermatomyositis with IgA nephropathy: case-based review Rheumatology International; 39: 577–581.
- 67. Sun C., Lee J.-H., Yang Y.-H. et al. (2015) Juvenile Dermatomyositis: A 20-year Retrospective Analysis of Treatment and Clinical Outcomes Chiang Pediatrics and Neonatology; 56: 31e39.
- 68. Ramanan A.V., Sawhney S. et al. (2001) Murray Central nervous system complications in two cases of juvenile onset dermatomyositis. Rheumatology, Volume 40, Issue 11: 1293–1298. doi.org/10.1093/rheumatology/40.11.1293.
- 69. Besnard C., Gitiaux C., Girard M. et al. (2019) Severe abdominal manifestations in juvenile dermatomyositis. BMJ, Volume 78, Issue Suppl. 2.
- 70. Dourado M.R., da Silva Filho T.J., Salo T. (2017) Oral signs in juvenile dermatomyositis J. Oral. Diag, 02: e20170027. 1–5. doi: 10.5935/2525–5711.20170027
- 71. Kang E.H., Lee E.B., Shin K.C. et al. (2005) Interstitial lung disease in patients with polymyositis, dermatomyositis and amyopathic dermatomyositis. Rheumatology (Oxford), 44: 1282–6.
- 72. Bowyer S.L., Blane C.E., Sullivan D.B. et al. (1983) Childhood dermatomyositis: factors predicting functional outcome and development of dystrophic calcification. J. Pediatr., 103: 882–8.
- 73. Національний підручник з ревматології (2013) За ред. В.В. Коваленка, Н.М. Шуби. 671 с.
- 74. Christopher-Stine L., Plotz P. H. (2004) Adult inflammatory myopathies. Best Practice and Research Clinical Rheumatology, Vol. 18, № 3: 331–344.
- 75. Varnier G.C., Rosina S., Ferrari C. et al. (2018) Development and Testing of a Hybrid Measure of Muscle Strength in Juvenile Dermatomyositis for Use in Routine Care Arthritis Care Res. (Hoboken). Sep; 70(9): 1312–1319. doi: 10.1002/acr.23491. Epub (2018) Aug 12.
- 76. Rider L.G., Werth V.P., Huber A.M. et al. (2011) Measures of adult and juvenile dermatomyositis, polymyositis, and inclusion body myositis. Arthritis Care Res. (Hoboken), Nov; 63, Suppl. 11: S118–57. doi: 10.1002/acr.20532. Review
- 77. Rider L.G., Aggarwal R., Machado P.M. et al. (2018) Update on outcome assessment in myositis. Nat. Rev. Rheumatol., May; 14(5): 303–318. doi: 10.1038/nrrheum.(2018).33. PMID: 29651119
- 78. Gao W., Yuan C., Zou Y. et al. (2020) Development and pilot testing a self-reported pediatric PROMIS app for young children aged 5–7 years. J. Pediatr. Nurs, 53: 74–83.
- 79. Patel R.E.V., Lai J.-S., Gray E. et al. (2019) Comparison of PROMIS computerized adaptive testing administered item banks vs fixed short forms in Juvenil myositis. Arthritis Rheum., 71(10): abstract # 433.
- 80. Rennebohm R.M., Jones K., Huber A.M. et al. (2004) Normal scores for nine maneuvers of the Childhood Myositis Assessment Scale. Arthritis Rheum., 51(3): 365–70.
- 81. Quinones R., Morgan G.A., Amoruso M. et al. (2013) Lack of achievement of a full score on the childhood myositis assessment scale by healthy four-year-olds and those recovering from juvenile dermatomyositis. Arthritis Care Res. (Hoboken). 65(10): 1697–701.
- 82. Aggarwal R., Rider L.G., Ruperto N. et al. (2017) International Myositis Assessment and Clinical Studies Group and the Paediatric Rheumatology International Trials Organisation. Ann. Rheum. Dis; 76: 78291.
- 83. Baechler E.C., Bauer J.W., Slattery C.A. et al. (2007) An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol. Med., 13: 59–68.
- 84. Ye Y., Fu Q., Wang R. et al. (2019) SerumKL-6 level is a prognostic marker in patients with anti-MDA5 antibody- positive dermatomyositis associated with interstitial lung disease. J. Clin. Lab. Anal., 33(8): e22978.
- 85. Vega P., Ibarra M., Prestridge A. et al. (2011) Autoantibody to PL-12 (anti-Alanyl-tRNA synthetase) in a African American girl with uvenile dermatomyositis and resolution of interstitial lung disease. J. Rheumatol., 38(2): 394–5.
- 86. Sabbagh S.P.-F.I., Kishi T. et al. (2019) Anti-Ro52 autoantibodies are associated with interstitial lung disease and more severe disease in patients with juvenile myositis. Ann. Rheum. Dis.; 78: 988–95. doi.org/10.1136/annrheumdis-2018–215004
- 87. Bohan A., Peter J.B. (1975) Polymyositis and dermatomyositis (first of two parts). N. Engl. J. Med., 292(7): 344–7.
- 88. Bohan A., Peter J.B. (1975) Polymyositis and dermatomyositis (second of two parts).N. Engl. J. Med., 292(8): 403–7.
- 89. Lundberg I.E., de Visser M., Werth V.P. (2018) Classification of myositis. Nature Reviews Rheumatology, volume14: 269–278.
- 90. Lundberg I.E., Tjärnlund A., Bottai M. et al. (2017) European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol.; 69(12): 2271–82.
- 91. Zhang X., Yang X., Ji L. et al. (2019) Validation of 2017 classification criteria for adult and juvenile idiopathic inflammatory myopathies proposed by EULAR/ACR in Chinese patients. Int. J. Rheum. Dis.; 22(7): 1278–82.
- 92. Jinnin M., Ohta A., Ishihara S. et al. (2020) First external validation of sensitivity and specificity of the European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria for idiopathic inflammatory myopathies with a Japanese cohort. Ann. Rheum. Dis.; 79(3): 387–92.
- 93. Huber A.M., Feldman B., Rennebohm R. et al. (2000) Validation and clinical significance of The Childhood Myositis Assessment Scale for assessment of muscle function in the juvenile idiopathic inflammatory myopathies. Arthritis Rheum., 50: 1595–1603.
- 94. Hashkes P.J., Wright B.M., Lauer M.S. et al. (2010) Mortality outcomes in pediatric rheumatology in the US. Arthritis Rheum., 62(2): 599–608.
- 95. Eimer M.J., Brickman W.J., Seshadri R. et al. (2011) Clinical status and cardiovascular risk profile of adults with a history of juvenile dermatomyositis. J. Pediatr., 159(5): 795–801.
- 96. Silverberg J.I., Kwa L., Kwa M.C. (2018) Cardiovascular and cerebrovascular comorbidities of juvenile dermatomyositis in US children: an analysisof the National Inpatient Sample. Rheumatology (Oxford); 57(4): 694–702.
- 97. Huber A.M., Kim S., Reed A.M. et al. (2017) Childhood Arthritis and Rheumatology Research Alliance Consensus clinical treatment plans for juvenile dermatomyositis with persistent skin rash. J. Rheumatol.; 44(1): 110–6.
- 98. Bellutti Enders F., Bader-Meunier B., Baildam E. et al. (2017) Consensus-based recommendations for the management of juvenile dermatomyositis. Ann. Rheum. Dis.; 76: 329–340.
- 99. Rouster-Stevens K.A., Gursahaney A., Ngai K.L. et al. (2008) Pharmacokinetic study of oral prednisolone compared with intravenous methylprednisolone in patients with juvenile dermatomyositis. Arthritis Rheum., 59(2): 222–6.
- 100. Feldman B.M., Rider L.G., Reed A.M. et al. (2008) Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet; 371(9631): 2201–12.
- 101. Ruperto N., Pistorio A., Oliveira S. et al. (2016) Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomized trial. Lancet, 387(10019): 671–8.
- 102. Klein-Gitelman M.S., Pachman L.M. (1998) Intravenous corticosteroids: adverse reactions are more variable than expected in children. J. Rheumatol.; 25(10): 1995–2002.
- 103. Giancane G., Lavarello C., Pistorio A. et al. (2019) The PRINTO evidence-based proposal for glucocorticoids tapering/discontinuation in new onset juvenile dermatomyositis patients. Pediatr Rheumatol. Online J., 17(1): 24.
- 104. Kishi T., Bayat N., Ward M.M. et al. (2018) Medications received by patients with juvenile dermatomyositis. Semin Arthritis Rheum.; 48(3): 513–22.
- 105. Shah M., Mamyrova G., Targoff I.N. et al. (2013) Rider, with the Childhood Myositis Heterogeneity Collaborative Study Group The Clinical Phenotypes of the Juvenile Idiopathic Inflammatory Myopathies Medicine, Volume 92: 25–41.
- 106. Maksimovic V., Pavlovic-Popovic Z., Vukmirovic S. et al. (2020) Molecular mechanism of action and pharmacokinetic properties of methotrexate. Mol. Biol. Rep., 47(6): 4699–708.
- 107. Papadopoli D.J., Ma E.H., Roy D. et al. (2020) Methotrexate elicits pro-respiratory and anti-growth effects by promoting AMPK signaling. Sci. Rep., 10(1): 7838.
- 108. Villarroel M.C., Hidalgo M., Jimeno A. (2009) Mycophenolate mofetil: an update. Drugs Today (Barc). 45(7): 521–32.
- 109. Rouster-Stevens K.A., Morgan G.A., Wang D. et al. (2010) Mycophenolate mofetil: a possible therapeutic agent for children with juvenile dermatomyositis. Arthritis Care Res. (Hoboken); 62(10): 1446–51.
- 110. Wang X., Wang H., Shen B. et al. (2016) 1-Alpha, 25-dihydroxyvitamin D3 alters the pharmacokinetics of mycophenolic acid in renal transplant recipients by regulating two extrahepatic UDP-glucuronosyltransferases 1A8 and 1A10. Transl. Res., 178: 54–62.e6.
- 111. Hassan J., van der Net J.J., van Royen-Kerkhof A. (2008) Treatment of refractory juvenile dermatomyositis with tacrolimus. Clin. Rheumatol.; 27(11): 1469–71.
- 112. Feng F., Li Y., Ji S. et al. (2019) Tacrolimus combined with corticosteroids effectively improved the outcome of a cohort of patients with immune-mediated necrotising myopathy. Clin. Exp. Rheumatol., 37(5): 740–7.
- 113. Chighizola C.B., Ong V.H., Meroni P.L. (2017) The use of cyclosporine a in rheumatology: a 2016 comprehensive review. Clin. Rev. Allergy Immunol., 52(3): 401–23.
- 114. Kishi T., Bayat N., Ward M.M. et al. (2018) Medications received by patients with juvenile dermatomyositis. Semin. Arthritis Rheum., 48(3): 513–22.
- 115. Deakin C.T., Campanilho-Marques R., Simou S. et al. (2018) Efficacy and safety of cyclophosphamide treatment in severe juvenile dermatomyositis shown by marginal structural modeling. Arthritis & rheumatology (Hoboken,NJ), 70(5): 785–93.
- 116. Hussein R.S., Khan Z., Zhao Y. (2020) Fertility preservation in women: indications and options for therapy. Mayo Clin. Proc.; 95(4): 770–83.
- 117. Ayza M.A., Zewdie K.A., Tesfaye B.A. et al. (2020) The role of antioxidants in ameliorating cyclophosphamide-induced cardiotoxicity. Oxidative Med. Cell. Longev.: 4965171.
- 118. Müller-Calleja N., Manukyan D., Canisius A. et al. (2017) Hydroxychloroquine inhibits proinflammatory signalling pathways by targeting endosomal NADPH oxidase. Ann. Rheum. Dis.; 76(5): 891–7.
- 119. Carter A.E., Eban R., Perrett R.D. (1971) Prevention of postoperative deep venous thrombosis and pulmonary embolism. Br. Med. J. 971; 1(5744): 312–4.
- 120. Rainsford K.D., Parke A.L., Clifford-Rashotte M. et al. (2015) Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology; 23(5): 231–69.
- 121. Das M., Karnam A., Stephen-Victor E. et al. (2020) Intravenous immunoglobulin mediates anti-inflammatory effects in peripheral blood mononuclear cells by inducing autophagy. Cell. Death Dis. 11(1): 50.
- 122. Angelotti F., Capecchi R., Giannini D. et al. (2020) Long-term efficacy, safety, and tolerability of recombinant human hyaluronidase facilitated subcutaneous infusion of immunoglobulin (Ig) (fSCIG; HyQvia (®)) in immunodeficiency diseases: real-life data from amonocentric experience. Clinical and experimental medicine. doi.org/10.1007/s10238–020–00633–4.
- 123. Chen Y., Wang C., Xu F. et al. (2019) Efficacy and tolerability of intravenous immunoglobulin and subcutaneous immunoglobulin in neurologic diseases. Clin. Ther., 41(10): 2112–36.
- 124. Fasano S., Alves S.C., Isenberg D.A. (2016) Current pharmacological treatment of idiopathic inflammatory myopathies. Expert Rev Clin. Pharmacol.; 9(4): 547–58.
- 125. Khojah A.M., Miller M.L., Klein-Gitelman M.S. et al. (2019) Rituximab-associated Hypogammaglobulinemia in pediatric patients with autoimmune diseases. Pediatr. Rheumatol. Online J.; 17(1): 61.
- 126. Oddis C.V., Reed A.M., Aggarwal R. et al. (2013) Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum., 65(2): 314–24.
- 127. Aggarwal R., Bandos A., Reed A.M. et al. (2014) Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheum. 66(3): 740–9.
- 128. Aggarwal R., Loganathan P., Koontz D. (2017) Cutaneous improvement in refractory adult and juvenile dermatomyositis after treatment with rituximab. Rheumatology (Oxford); 56(2): 247–54.
- 129. Khoo T., Limaye V. (2020) Biologic therapy in the idiopathic inflammatory myopathies. Rheumatol. Int. 40(2): 191–205.
- 130. Rouster-Stevens K.A., Ferguson L., Morgan G. et al. (2014) Pilot study of etanercept in patients with refractory juvenile dermatomyositis. Arthritis Care Res. (Hoboken); 66(5): 783–7.
- 131. Brunasso A.M., Aberer W., Massone C. (2014) New onset of dermatomyositis/polymyositis during anti-TNF-α therapies: a systematic literature review. The Scientific World Journal: 179180.
- 132. Wenhong J., Zhang Z., Li, Y. et al. (2021) The Cell Origin and Role of Osteoclastogenesis and Osteoblastogenesis in Vascular Calcification Front. Cardiovasc. Med., 23 Apri. doi: org/10.3389/fcvm.2021.639740
- 133. Lorenzetti R., Janowska I., Smulski C.R. et al. (2019) Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J. Autoimmun., 101: 145–52.
- 134. Papadopoulou C., Hong Y., Omoyinmi E. et al. (2019) Janus kinase 1/2 inhibition with baricitinib in the treatment of juvenile dermatomyositis. Brain., 142(3): e8.
- 135. Smith L.N., Paik J.J. (2020) Promising and Upcoming Treatments in Myositis. 2 Curr. Rheumatol. Rep.; Aug 26; 22(10): 65. Published online (2020) Aug 26. doi: 10.1007/s11926–020–00943–2 Lenabasum progresses to Phase 3 clinical trials July 25, 2018.
- 136. Sanner Н. (2014) Disease activity and prognostic factors in juvenile dermatomyositis: a long-term follow-up study applying the Paediatric Rheumatology International Trials Organization criteria for inactive disease and the myositis disease activity assessment tool/Helga Sanner, Ivar Sjaastad, Berit Flatø. Oxford Journals Rheumatology; № 2 (29). 10.1093/rheumatology/keu146.
- 137. Rider L.G., Aggarwal R., Pistorio A. et al. (2016) International Myositis Assessment and Clinical Studies Group and the Paediatric Rheumatology International Trials Organisation. (2016) American College of Rheumatology/European League Against Rheumatism Criteria for Minimal, Moderate, and Major Clinical Response in Juvenile Dermatomyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative.
- 138. American College of Rheumatology/European League Against Rheumatism criteria for minimal, moderate, and major clinical response in adult dermatomyositis and polymyositis (2017) An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann. Rheum. Dis.; 76: 792801.
Ювенильный дерматомиозит. О чем следует помнить взрослому ревматологу?
1Национальный университет охраны здоровья Украины имени П.Л. Шупика
2ГУ «Институт педиатрии, акушерства и гинекологии имени академика Е.М. Лукьяновой НАМН Украины»
3Харьковский национальный университет имени В.Н. Каразина
4ГУ «Институт охраны здоровья детей и подростков НАМН Украины»
5ГУ ННЦ «Институт кардиологии имени акад. М.Д. Стражеско» НАМН Украины
Резюме. Обоснование. Ювенильный дерматомиозит (ЮДМ) — тяжелое редкое прогрессирующее системное заболевание аутоиммунного генеза с преимущественным поражением поперечно-полосатых мышц, кожи и сосудов микроциркуляторного русла, которое развивается у детей до 16-летнего возраста, но, несмотря на тяжесть, смертность от ЮДМ значительно ниже, чем у пациентов взрослого возраста с дерматомиозитом/полимиозитом, также вдвое реже отмечается инвалидизация пациентов детского возраста. ЮДМ имеет определенные иммунологические и клинические различия в развитии и течении дерматомиозита у детей по сравнению с лицами взрослого возраста. Методы исследования. На основании литературных данных в статье поданы современные представления относительно этиологии и патогенеза, описаны диагностические критерии ЮДМ; проведено сопоставление клинических и лабораторных проявлений и частоты их выявления у пациентов детского и взрослого возраста, подчеркнуты основные отличия дифференциальной диагностики. Приведены унифицированные терапевтические стратегии согласно рекомендациям международных ревматологических ассоциаций. Выводы. Учитывая текущие терапевтические возможности, пациенты с ЮДМ составляют все большую долю среди пациентов с воспалительными миозитами. Это обосновывает необходимость дальнейших исследований менеджмента больных с ЮДМ для улучшения уровня оказания медицинской помощи и отдаленных последствий болезней.
Ключевые слова: дети, дерматомиозит, особенности.
Адреса для листування:
Ошлянська Олена Анатоліївна
E-mail: eoshljanskaja17@gmail.com
Leave a comment