РІДКІСНІ ХВОРОБИ: ДВА ВИПАДКИ ПРОГРЕСУЮЧОЇ ОСИФІКУЮЧОЇ ФІБРОДИСПЛАЗІЇ У ПРАКТИЦІ ДИТЯЧОГО РЕВМАТОЛОГА
Ошлянська О.А.1, Арцимович А.Г.2, Самоненко Н.В.3
- 1Національна медична академія післядипломної освіти ім. П.Л. Шупика МОЗ України, Київ
- 2ДУ «Інститут педіатрії, акушерства і гінекології ім. акад. О.М. Лук’янової НАМН України», Київ
- 3НДСЛ «Охматдит», Київ
Резюме. Прогресуюча осифікуюча фібродисплазія є рідкісним (1 на 2 млн) генетичним захворюванням з аутосомно-домінантним типом успадкування, яке полягає у поступовому заміщенні м’язової тканини кістковою і призводить до інвалідизації, а згодом смерті пацієнта. Шлях до встановлення діагнозу зазвичай займає декілька років у зв’язку з низькою розповсюдженістю патології та клінічною подібністю до ортопедичних чи онкологічних захворювань. На сьогодні ця хвороба є невиліковною, але у світі тривають клінічні випробування препаратів, що дозволять призупинити процес осифікації. Мета: привернути увагу до особливостей перебігу і труднощів діагностики прогресуючої осифікуючої фібродисплазії. Об’єкт: пацієнти педіатричного профілю з прогресуючою осифікуючою фібродисплазією. Методи: опрацювання даних медичної документації. Результати: описані два випадки прогресуючої осифікуючої фібродисплазії у дітей, які перебували на лікуванні у клініці ДУ «ІПАГ ім. акад. О.М. Лук’янової НАМН України». Висновки: настороженість дитячих ревматологів та лікарів суміжних спеціальностей щодо виявлення випадків прогресуючої осифікуючої фібродисплазії дозволить якомога раніше надавати всю можливу допомогу пацієнтам із цим діагнозом та поліпшувати їхню якість життя.
УДК 616-039.42:616-002.77
DOI: 10.32471/rheumatology.2707-6970.78.13031
Однією із патологій, шлях до діагностики якої є зазвичай дуже довгим, а прогноз невтішним, є прогресуючий осифікуючий міозит або прогресуюча осифікуюча фібродисплазія, хвороба, що перетворює людей на «живі статуї». Назва хвороби походить від лат. Fibrō «волокно» + dis «розлад, порушення» + грец. Πλάσις «будова, структура» (лат. Os, ossis «кістка» + facio «робити» = «окостеніння») і означає, що «м’яка сполучна тканина прогресивно перетворюється в кістку». Така назва запропонована в 1970-х роках батьком сучасної медичної генетики доктором В. МакКьюзіком.
Загалом, як відомо, осифікація (остеогенез, окостеніння) — це процес формування кісткової тканини, який в організмі є вкрай важливим для росту дитини та процесів відновлення кісткової тканини після різних ушкоджень. Гетеротопічною осифікацією називають утворення кісткової тканини у нетипових місцях (позаскелетна осифікація).
До осифікуючих міозитів відносять цілу групу захворювань сполучної тканини, при яких м’язова тканина заміщується кістковою: травматичний осифікуючий міозит, прогресуючий осифікуючий міозит і трофоневротичний осифікуючий міозит. До травматичних осифікуючих міозитів включені набуті внаслідок травми запальні захворювання м’язів і зв’язок, які характеризуються патологічним окостенінням у травмованому місці чи поруч з ним. Осифікуючий міозит уражає глибокі відділи скелетних м’язів поблизу окістя, яке при цьому буває різко потовщене. Найчастіша локалізація — стегна, плечі та сідниці. Мікроскопічна картина його складається з геморагії, організованої гематоми, хрящових формацій, енхондральної та періостальної осифікації. Трофоневротичний, або нейротрофічний, міозит розвивається після перенесеної травми спинного мозку, великих нервових стовбурів і зазвичай проявляється в тазостегновому (кульшовому) і колінному суглобах (Favus M.J., 1990).
На відміну від них, прогресуюча осифікуюча фібродисплазія (fibrodisplasia ossificans progressiva) розвивається в ранньому дитячому віці і є генетично зумовленою, рідкісною хворобою, що характеризується повторними епізодами припухлості м’яких тканин, їх гетеротопною мінералізацією з перетворенням сполучної тканини у м’язах, сухожиллях і зв’язковому апараті у кісткову тканину.
Перший опис її належить Juy Patin (1692). У 1869 р. Mimchmeyer систематизував 12 випадків, тому хвороба отримала його ім’я. Зараз у МКХ-10 вона розглядається за кодом М61.1 і внесена до списку орфанних захворювань (orphanet), створено окреме товариство з її вивчення (The International Fibrodysplasia Ossificans Progressiva Association — IFOPA), а з 1988 р. існує Міжнародна організація хворих на фібродисплазію (www.ifopa.org).
Етіологія прогресуючої осифікуючої фібродисплазії досі залишається недостатньо вивченою. З 1960-х років обговорюється роль вродженого порушення остеогенезу. Вважається, що це ембріогенетична аномалія, яка супроводжується порушенням у міжм’язовій сполучній тканині внаслідок недостатнього диференціювання мезенхіми з аномальною властивістю виробляти кісткову тканину. На користь цієї теорії свідчить як розвиток дебюту цієї хвороби в дитячому віці, так і підвищення частоти різних вроджених дефектів (мікродактилія перших пальців кистей і стоп, аномалії розвитку хребта, гіпогонадизм, глухота; можлива відсутність мочок вуха або різців). На думку S. Mahboubi та співавторів (2001), прогресуюча осифікуюча фібродисплазія є генетичним захворюванням, що успадковується домінантно з варіабельною експресією і повною пенетрантністю. Передбачається, що мутація в гені ACVP1/ALR2 (Lucotte G., Bathelier C. et al., 2000; Semonin O. et al., 2001) спричиняє синтез аномального рецептора (R206H) чи гіперпродукції морфогенного білка кістки (bone morphogenetic protein-4) (Hegyl L., Gannon F.H., 2003; Glaser D.L. et al., 2003). Внаслідок цього міобласт перетворюється на остеобласт (Olmsted E.A., Kaplan F.S., 2003). Новітні дослідження свідчать, що розвиток фібродисплазії зумовлений порушеннями активінзалежної передачі сигналу у фібро-/адипогенних клітинах-попередниках за відсутності у дитини алеля ACVR1, внаслідок чого в організмі різко загострюється гетерогенна осифікація. Це дозволяє припустити, що дикі та мутантні ACVR1-рецептори конкурують за ліганди активіну або зв’язувачі рецепторів кісткового морфогенетичного білка ІІ типу (Lees-Shepard J.B. et al., 2018). Деякими вченими висувається гіпотеза про вірусну етіологію хвороби (Scarlett R.F. et al., 2004). Так, І. Гаусманова-Петрусевич (1971) вважає прогресуючу осифікуючу фібродисплазію однією з форм універсального кальцинозу. Також висловлюються припущення на користь запальної природи захворювання чи його ендокринного походження (зокрема ураження надниркових залоз і паращитоподібних залоз).
Поширеність хвороби становить близько 1 на 2 млн людей; не відзначається статевої, расової та етнічної схильності.
На першому році життя патогномонічною ознакою прогресуючої осифікуючої фібродисплазії є лише вальгусна деформація та мікродактилія І пальця стоп (клінодактилія), яка відзначається майже у всіх пацієнтів (Mahboubi S., Kaplan F.S., 2001; Blaszczyk M. еt al., 2003).
Поступово до клінічної картини додаються ектопічні осифікати. Їх прояви маніфестують у віці 3–4 роки появою припухлості тканин в ділянці шиї, спини або плечового поясу, з’являється набряклість, нерідко болючість, місцева гіперемія, гіпертермія та лихоманка. Щільність ураженого вогнища спочатку підвищується, а потім поступово зникає і супроводжується затвердінням м’яза. Проте не всі ущільнені вогнища надалі осифікуються. Спочатку ущільнення можуть бути рентгенологічно неконтрастними.
Характерним є хвилеподібний перебіг хвороби, інтервали між загостреннями можуть бути досить великі. Водночас захворювання неухильно прогресує, захоплюючи все більшу частину опорно-рухового апарату, що перетворює хворих у «скам’янілих людей». Найбільш схильні до окостеніння місця прикріплення м’язів до кісток, менше — їх центральні відділи. Часто уражаються поверхневі м’язи, головним чином задньої поверхні тулуба і кінцівок. Практично всіма авторами відзначається тенденція до утворення стійких згинальних контрактур, м’язових атрофій, обмеження рухливості хребта, а при ураженні груднинно-ключично-соскоподібного м’яза — розвиток кривошиї. Аномалії шийного відділу хребта — периферичні остеохондроми з подальшою осифікацією і зрощенням — розвиваються у дітей старшого віку (до 90% пацієнтів) (Palhares D.B., Leme L.M., 2001).
При прогресуванні хвороби може порушуватися процес жування, тому суттєво утруднюється харчування хворих; відзначають деформацію грудної клітки, що призводить до розвитку частих рестриктивних легеневих захворювань і пневмоній. Осифікати можуть утворюватися майже у всіх тканинах і органах тіла людини: в рубцево-зміненій шкірі, у скелетних м’язах, сухожиллях, у жировій клітковині, у зв’язках і суглобовій капсулі.
Характерний зовнішній вигляд хворого: хода скута, голова нахилена дещо вперед, обличчя амімічне, м’язи шиї мають вигляд натягнутих тяжів, різко обмежені рухи в усіх відділах хребта. Не описані осифікати лише в м’язах язика, м’якого піднебіння, глотки, діафрагми, сфінктерів, серця і в гладких м’язах. Деякі автори відзначали супутню патологію ендокринних залоз, яєчників, атрофію яєчок, ураження надниркових і щитоподібної залоз (Пономарев А.А., 1998).
На думку F.S. Kaplan та співавторів (2005), у 95% хворих початок осифікації м’язів також провокується травмою. Патогномонічними вважаються екзостози на широкій ніжці.
Мюнхмайер і Ніколадоні розрізняють три стадії розвитку окостеніння:
I стадія (інфільтрації) — розростання молодої дегенеративної тканини і вторинні дегенеративні зміни в м’язах. Рентгенологічно ці зміни не визначаються, а гістологічне обстеження виявляє запальні зміни в міжм’язовій сполучній тканині з її набряком, круглоклітинною інфільтрацією і появою кистоподібних утворень.
II стадія (фіброзної індурації) — відбувається ущільнення сполучної тканини, її рубцювання із вторинною атрофією м’язової тканини. Рентгенологічно виявляються «ніжні» тіні за типом кісткової мозолі.
III стадія (окостеніння): утворення кісткової тканини в місцях ураження м’яких тканин, що чітко проявляється на рентгенограмах інтенсивними тінями.
Слід зауважити, що всі стадії хвороби можуть розвиватися одночасно у різних відділах скелета.
Патоморфологічна картина свідчить про те, що за своєю будовою новостворена кістка нічим не відрізняється від нормальної. Вогнища кісткової тканини поступово збільшуються, зливаються між собою, набуваючи гіллястої форми з губчастої речовини в глибині та компактної форми — у поверхневих відділах кістки. У старіших кісткових утвореннях серед балок з’являється кістковий мозок.
Розрізняють два типи кісткової метаплазії у хворих на прогресуючу осифікуючу фібродисплазію. При першому спостерігається розпушування сполучної тканини, збільшення кількості ядер; відкладення солей кальцію серед волокон сполучної тканини і трансформація клітин безпосередньо в кісткові тільця без/з появою типових остеобластів. Другий тип характеризується попереднім утворенням хряща (Проскурова В.И., Костенко И.Н., 1973).
Рентгенологічна картина зазвичай представлена двома чи трьома формами, описаними вище: тіні вільні, ще не кісткової інтенсивності, кісткові сформовані утворення, не пов’язані з кістками скелета, та екзостози. Рентгенологічна картина залежить від фази патологічного процесу: спочатку видно тіні фіброзно ущільнених поєднаних прошарків м’язів, пізніше з’являються ділянки кісткової щільності ,та в подальшому визначаються деталі кісткової структури.
При комп’ютерній томографії візуалізується кальцифікація гетеротопічної кістки, яка просувається від зовнішнього краю першого вузла до центру. Радіоізотопне дослідження дозволяє виявити активний міозит з інтенсивним позакістковим накопиченням ізотопів.
При патологоанатомічному дослідженні визначається атрофія скелетних м’язів і заміщення їх фіброзною тканиною, окостеніння м’язів, зв’язок, сухожиль, апоневрозів, екзостози, гіперостоз, потовщення кісток із розпушуванням губчастої речовини епіфізів за відсутності змін у суглобах.
При лабораторному обстеженні можуть виявлятися ознаки метаболічного ацидозу, анемії. Гострофазової відповіді, вираженої запальної реакції чи порушення кальцієвого балансу зазвичай не відзначається. Не характерне також підвищення вмісту креатинфосфокінази, що дозволяє відрізняти прогресуючу осифікуючу фібродисплазію від запальних міопатій.
Диференційний діагноз на початкових стадіях хвороби проводять з іншими гетеротопними окостеніннями, первинним кальцинозом, паразитарними інвазіями (цистицеркозом тощо), новоутвореннями, дефіцитом вітаміну D, післяін’єкційними гранульомами.
І.П. Нікішина (Антелава О.А. и соавт., 2015) припускає, що прогресуюча осифікуюча фібродисплазія за перебігом може наближатися до ревматичних хвороб, про що свідчить висока частота розвитку псоріазу, синовіту, сакроілеїту при прогресуючій осифікуючій фібродисплазії.
Достовірних відомостей про можливість припинення хвороби або ефективної фармакотерапії прогресуючої осифікуючої фібродисплазії немає.
Операційне видалення осифікатів вважається протипоказаним, оскільки можлива провокація ще більшого поширення патологічного процесу. З того ж приводу не використовується ортопедична корекція.
Ефективність глюкокортикоїдів не доведена, хоча в дебюті хвороби використовується пульс-терапія. Знеболювальний ефект досягається тривалим призначенням нестероїдних протизапальних препаратів (НПЗП). Розглядається можливість використання антицитокінової терапії (анти-ФНП), продовжуються клінічні дослідження агоністів ретиноїдної кислоти та штучних антитіл до активіну А. З 2014 р. розпочата друга фаза двох клінічних досліджень препарату паловаротен (Clementia Pharmaceuticals, 2014).
Також пацієнтам призначають етилендіамінтетраоцтову кислоту (ЕДТА), калію йодид, вітаміни групи В, С, А, біостимулятори. При цьому слід уникати застосування внутрішньом’язових ін’єкцій, які можуть спровокувати утворення нових вогнищ осифікації.
Прогноз при прогресуючій осифікуючій фібродисплазії досі вважається безнадійним. Найчастіші причини летальності — легенева інфекція на тлі гіповентиляції внаслідок ураження міжреберних м’язів, виснаження, яке зумовлене окостенінням жувальних і ковтальних м’язів. Проте описані й поодинокі приклади спонтанного регресу хвороби.
Незважаючи на рідкість захворювання, протягом останніх кількох років у нашій практиці було два випадки, які наводимо.
Випадок 1
Дівчинка Ж., 5 років. Ранній анамнез не обтяжений, хворіла зрідка. У сім’ї — 9 дітей. Інші діти, зі слів, здорові, не оглянуті. Родина соціально неблагополучна. Мати вважає дитину хворою з 5 років (із серпня 2015 р.), коли вперше помітила припухлість біля правого кута лопатки, що була болісна при пальпації (рис. 1). З цими скаргами була госпіталізована за місцем мешкання, де дитину обстежено.

У загальному аналізі крові, сечі, біохімічному аналізі крові змін не виявлено. Показники АНА, анти-ЦЦП-антитіла, анти-ДНК-ІІ-антитіла — негативні, субпопуляції лімфоцитів, вміст імуноглобулінів — в межах нормальних значень.
ЕКГ, УЗД органів черевної порожнини — без особливостей.
Встановлений діагноз: Пухлина м’яких тканин? Недиференційоване системне захворювання сполучної тканини?
Проведено КТ органів грудної клітки: ознаки помірно вираженого пневмофіброзу. Об’ємно-інфільтративні зміни м’яких тканин правої бічної стінки грудної клітки. Ділянки патологічного кісткового утворення у м’яких тканинах лівої передньої грудної стінки. Для подальшого обстеження була направлена в ДУ «ІПАГ ім. академіка О.М. Лук’янової НАМН України» (Київ).
При госпіталізації стан дитини задовільний, скарг активно не пред’являє. Водночас під час огляду не може підняти руки до рівня пліч (див. рис. 1), рот відкриває частково, повертається всім тулубом разом із шиєю. М’язи шиї, спини, плечового поясу ущільнені, більше ліворуч, проте пальпація чутливіша праворуч. Там же біля кута лопатки візуалізується набряклість м’яких тканин, щільне утворення. Рухи у суглобах, окрім плечових та ліктьових — не обмежені. Ознак активного синовіту немає. Слизова оболонка та шкіра чисті. З боку внутрішніх органів без суттєвих особливостей. Фізіологічні відправлення не порушені.
У стаціонарі дитина всебічно обстежена:
Загальний аналіз крові: еритроцити — 4,78·1012/л, гемоглобін — 120 г/л, лейкоцити — 7,2·109/л, тромбоцити — 342·109/л, еозинофіли — 6%, лімфоцти — 40%, паличкоядерні нейтрофіли — 3%, сегментоядерні нейтрофіли — 46%, моноцити — 5%, ШОЕ — 15 мм/год.
Біохімічне дослідження крові: загальний білірубін — 6,6 мкмоль/л, тимолова проба — 2,0 Од, АлАТ — 19 О/л, АсАТ — 21 О/л, загальний білок — 73,3 г/л, сечовина — 4,02 мкмоль/л, креатинін — 0,052 мкмоль/л, глюкоза — 3,81 мкмоль/л, СРБ — негативний.
КФК — 177,0 Од/л, кальцій — 1,63 ммоль/л, фосфор — 1,60 ммоль/л, лужна фосфатаза загальна — 1552 Од/л, кишкова фракція — 428,9 (27,6%), кісткова фракція — 859,4 (55,1%).
Нормальний вміст ферментів міолізу дозволив виключити запальні та незапальні міопатії.
Кальцій у добовій сечі: 1,8 ммоль/д, фосфор — 10,5 ммоль/д (знижений).
Для виключення первинного гіперпаратиреозу досліджено паратгормон — 18,5 пг/мл.
Коагулограма: протромбіно-тромбіновий індекс — 82%, час рекальцифікації — 95 с, фібриноген загальний — 4,42 г/л, фібриноген «В» — (+++).
Імунологічне дослідження: IgG — 8,1 г/л, IgA — 0,79 г/л, IgM — 0,6 г/л, антитіла до посмугованих м’язів не виявлені. ЦІК — 0,025 г/л (н), AНA-негативні.
Загальний аналіз сечі, копрологічне дослідження — без особливостей.
ЕКГ: синусова тахікардія. ЧСС — 111 уд./хв. Відхилення електричної осі серця ліворуч — 1 градус. Неповна блокада правої ніжки пучка Гіса. Помірні обмінні зміни в міокарді.
УЗД органів черевної порожнини: без особливостей. Розміри паренхіматозних органів не збільшені.
ЕхоКГ: аномальна хорда лівого шлуночка. Розміри порожнин серця не збільшені. Гіпертрофії стінок немає. Скоротливість міокарда задовільна.
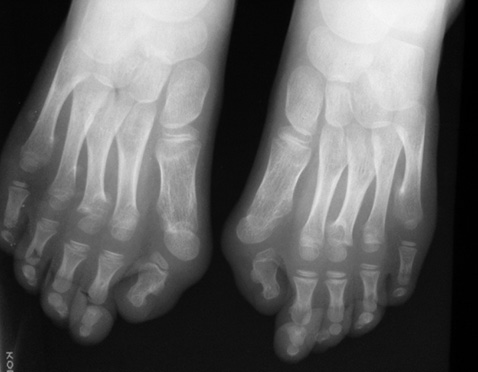
У зв’язку з виявленням клінодактилії мізинців стоп (рис. 2) проведена рентгенографія стоп: деформуючий поліартроз (?).
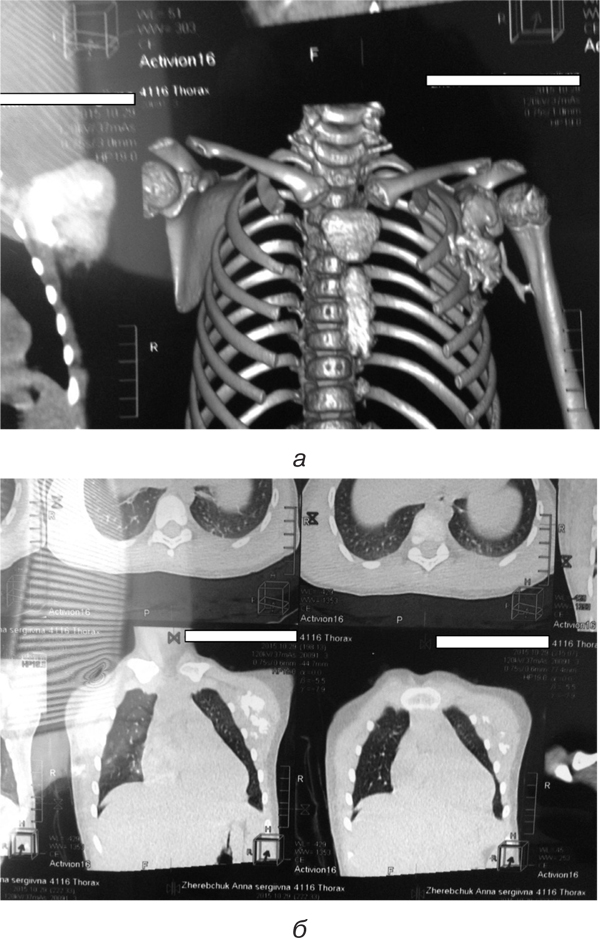
Магнітно-резонансна томографія (МРТ) органів грудної клітки: кальцифіковані утворення у м’язах (рис. 3).
Остеосцинтиграфія: виявляється помірно підвищена фіксація РФП в ділянці верхнього краю лівої лопатки — до 117%. Решта симетричних ділянок кісткової системи РФП фіксує рівномірно. Зазначене свідчило, що м’язова тканина накопичує кальцій тою ж мірою, як і кістки.
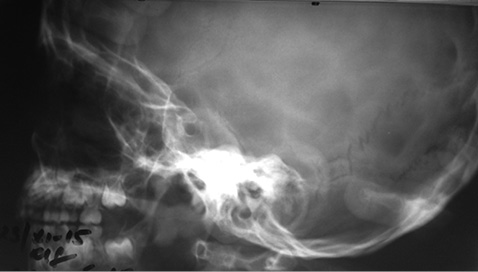
Рентгенографія черепа (рис. 4) проведена для виключення ендокраніальних кальцинатів, характерних для протозойних і паразитарних інвазій: «турецьке сідло» правильної форми.
Дитина консультована спеціалістами: окулістом (без патології), ЛОР (без патології), неврологом (вогнищевої неврологічної симптоматики не виявлено), онкологом в Національному інституті раку (виключено злоякісну пухлину).
Оскільки не вдалося знайти можливості для генетичного дослідження, проведено біопсію м’язів плеча та спини. Гістологічне дослідження тканини біоптату: у біоптаті з кута лівої лопатки наявні вогнища жирових клітин, сполучнотканинних структур із судинними пучками і набряком. Запальна інфільтрація не виявлена. В частині з них виявлені ділянки дрібновогнищевої мальформації судин венозного та артеріального типів з облітерацією. Множинні вогнищеві скупчення фібробластоподібних клітин з акантозом.
Отримані результати обстеження (наявність типової клінічної картини і відповідних стигм дизембріогенезу, змін кальцій-фосфорного обміну, відсутність ознак міолізу, загальнозапальної реакції, патогномонічні результати остеосцинтиграфії та гістологічного дослідження) дозволили встановити діагноз: прогресуюча осифікуюча фібродисплазія (прогресуючий осифікуючий міозит) (М61.1).
Як симптоматичне лікування дитина отримувала ібупрофен, електрофорез з КІ на праву лопаткову ділянку. Проведено бесіду з матір’ю про особливості перебігу і прогноз захворювання.
Дані рекомендації: спостереження в дільничного педіатра, ортопеда, продовжити прийом ібупрофену до 3–4 міс, повторні курси етидронової кислоти, курси електрофорезу з розсмоктувальними засобами.
На повторний огляд батьки дитину не привезли. Катамнез невідомий. Прогноз загалом несприятливий.
Випадок 2
Дівчинка Г., 7 років. Зі слів батьків росла та розвивалася відповідно до віку. Зі слів бабусі відомо, що сімейний анамнез обтяжений: батько дитини «також погано рухається». Була госпіталізована на початку вересня 2019 р. у Центральну дитячу міську лікарню за місцем проживання зі скаргами на набряк, біль та обмеження згинання в лівому колінному суглобі, що вперше з’явилися через деякий час після падіння з самоката у липні 2019 р. Госпіталізована за місцем проживання, де встановлено діагноз «реактивний артрит», відзначалося короткочасне підвищення ШОЕ до 20 мм/год. За даними інструментальних обстежень (рентгенографія та УЗД), проведених за місцем проживання, описані утворення за типом екзостозів у ділянці обох колінних суглобів. Отримували НПЗП коротким курсом, ефект частковий. У жовтні 2019 р. у дитини відзначений повторний набряк лівого колінного суглоба, з приводу чого дівчинка була повторно госпіталізована, під час обстеження за місцем проживання проведені дослідження АНА, HLA B27, антитіл до циклічного цитрулінованого пептиду, антитіл до хламідійних антигенів, які всі були негативні, за даними УЗД та МРТ виявлено синовіт лівого колінного суглоба, зберігалося також незначне підвищення ШОЕ. Дитина була направлена до ДУ «ІПАГ ім. акад. О.М. Лук’янової НАМН України» з діагнозом «ювенільний ідіопатичний артрит?» для уточнення діагнозу та визначення тактики терапії.
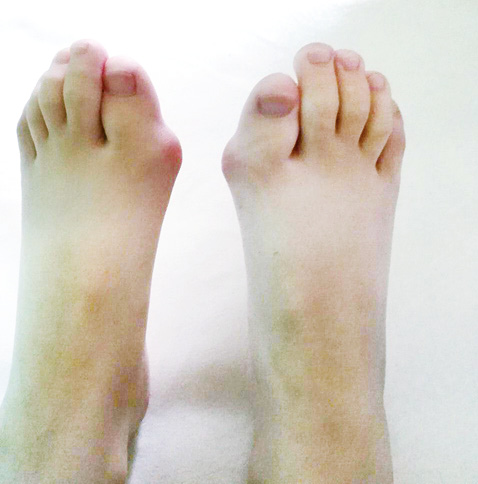
При первинному фізикальному огляді виявлено різкі обмеження поворотів шиї праворуч та ліворуч, повністю неможливе закидання голови назад (зі слів родичів, у батька дитини так само різко обмежені рухи шиї, чи обстежений батько з приводу цього — невідомо, з родиною не проживає), обмеження відведення нижньої щелепи, тугорухливість променезап’ясткових суглобів, деформації обох перших пальців стоп (рис. 5), набряк, локальна гіпертермія та обмеження згинання лівого колінного суглоба, при спробі виконати повне пасивне згинання дитина скаржилася на сильний біль. Інші суглоби при госпіталізації були без набряків та локальної гіпертермії. Аускультативно відзначалося дещо приглушене везикулярне дихання над легенями. З боку інших внутрішніх органів під час огляду змін не виявлено. Під час госпіталізації у дитини спостерігалися короткочасні епізоди набряків міжфалангових суглобів пальців кистей, що самостійно минали.
Проведені загальноклінічні обстеження були без істотних відхилень:
Загальний аналіз крові: гемоглобін — 129 г/л, еритроцити — 4,98·1012/л, лейкоцити — 7,86·109/л, тромбоцити — 465·109/л, ШОЕ — 3 мм/год, еозинофіли — 4,6%, нейтрофіли — 49,9%, лімфоцити — 34,5%, моноцити — 9,4%, б. 1,3%.
Біохімічний аналіз крові: білірубін — 9,3 мкмоль/л, холестерин — 3,4 мкмоль/л, тимолова проба — 3,0 Од, лужна фосфатаза — 234 од/л, АлАТ — 27 од/л, АсАТ — 32 од/л, сечовина — 4,39 мкмоль/л, загальний білок — 77,3 г/л, креатинін — 0,075 ммоль/л, глюкоза — 4,32 ммоль/л.
Коагулограма: ПТІ — 76,0%, фібриноген — 4,11 г/л, фібрин — 19 мг, фібриноген В — негативний.
Імунологічні дослідження: у сироватці крові імуноглобулін G — 15 г/л, імуноглобулін А — 2,3 г/л, імуноглобулін М — 1,75 г/л.
Загальний аналіз сечі та копрограма без відхилень. У добовій сечі глюкоза та білок не виявлені.
ЕКГ: синусовий ритм. ЧСС — 79 уд./хв. Нормальне положення електричної вісі серця. ЕКГ в межах норми.
УЗД щитовидної залози ознак ехоструктурних змін не виявило.
За даними УЗД ОЧП та нирок візуалізуються мезентеріальні лімфатичні вузли розміром 10–12 мм звичайної структури, реактивні зміни паренхіми печінки, тканини підшлункової залози, відзначена пієлоектазія обох нирок.
Враховуючи відсутність типових ознак та лабораторних змін, притаманних ювенільному ідіопатичному артриту, продовжували діагностичний пошук.
За даними інструментальних обстежень:
Спірографія: легке порушення вентиляційної здатності за рестриктивним типом. Дуже легке порушення вентиляційної здатності за обструктивним типом.
МРТ лівого стегна (рис. 6): осередків патологічної зміни МР-сигналу від м’яких тканин видимих ділянок правого та лівого стегна не виявлено. М’язи стегна та їх сухожилля сформовані правильно. Осередків обмеження дифузії на DW1 на досліджуваному рівні не виявлено. Видимі відрізки стегнових кісток виглядають інтактними, цілісність кортикального шару не порушена. У супрапателярній сумці та в порожнині лівого колінного суглоба визначається патологічне накопичення рідини. МР-ознак об’ємних, травматичних та інфільтративно-запальних змін структур лівого стегна при цьому обстеженні не виявлено. МР-картина синовіту лівого колінного суглоба.
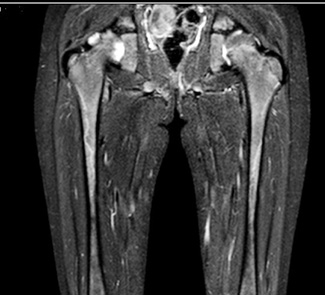
УЗД кистей: УЗ-ознаки ексудативного синовіту п’ястково-фалангового суглоба 3-го пальця правої кисті. УЗ-ознаки тендосиновіту сухожилля згинача 3-го пальця правої кисті.
УЗД ліктьових суглобів: УЗ-ознаки незначного синовіту ліктьових суглобів.
УЗД колінних суглобів: УЗ-ознаки зниження хряща, ексудативно-проліферативного синовіту лівого колінного суглоба.
УЗД гомілковостопних суглобів УЗ-патології м’яких тканин гомілковостопних суглобів не виявлено.
УЗД кульшових суглобів: УЗ-ознаки можуть відповідати остеофіту правого кульшового суглоба (гіперехогенне включення до 3 мм справа).
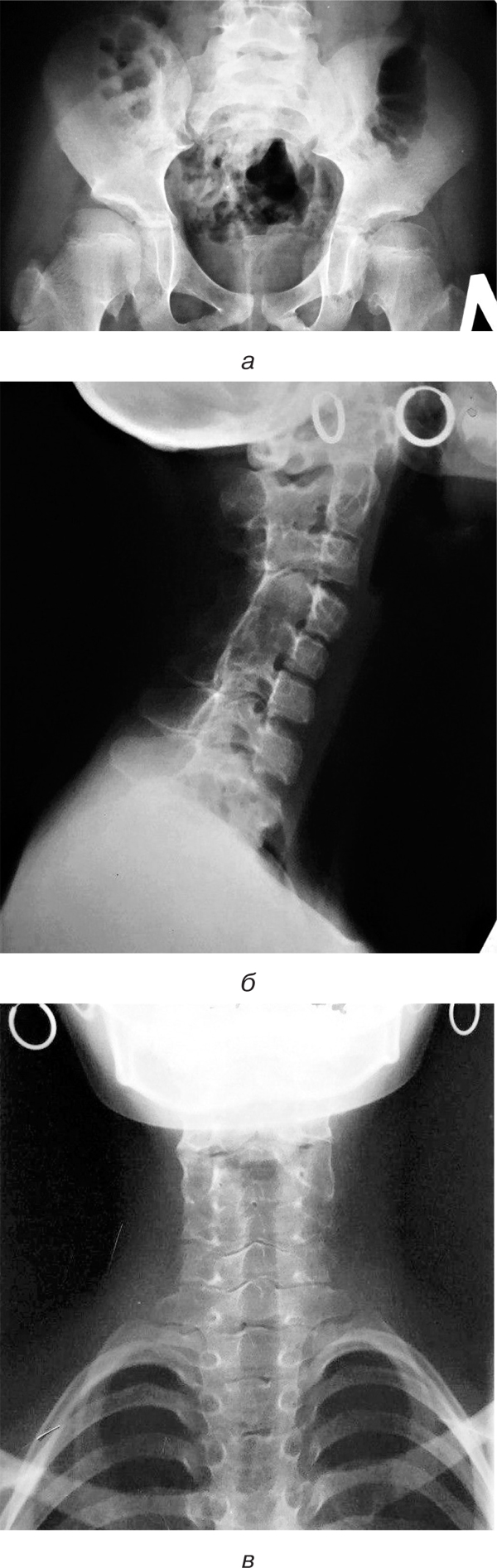
Рентгенографія кульшових суглобів. Суглобові западини мають нерівну поверхню. Суглобові щілини звужені зі зниженою прозорістю. Головки стегнових кісток виходять за межі западин (рис. 7а).
Рентгенографія шийного відділу хребта. Остисті відростки всіх шийних хребців зрощені. СІ– та СІІ-тіла хребців зрощені. Між іншими хребцями шиї мінімальний просвіт (рис. 7б, в).
Дитина отримала консультацію в ортопеда ДУ «Інститут травматології та ортопедії НАМН України», описана як вроджена екзостозна хвороба (діафізарна аклазія).
З огляду на обтяженість сімейного анамнезу виявлені зміни на рентгенограмі кульшових суглобів, характерні вроджені деформації великих пальців обох стоп, запідозрена вроджена системна патологія сполучної тканини, дитину направлено на консультацію до медичного генетика.
За тонкошаровою хроматографією амінокислот і вуглеводів суттєвих відхилень не виявлено: позитивні: тест на фруктозу, гіпераміноацидурію, пролін, сліди: на цистин, на мукополісахариди. Метаболіти сполучної тканини в добовій сечі: оксипролін 1062 ммоль/добу (норма — 191–488), рівень кислих ГАГ 100 од ЦПХ/г креатиніну (норма — 173 од ЦПХ/г креатиніну). Проведений біохімічний скринінг на мукополісахаридози І та ІІ типу методом сухої краплі.
Оскільки дані результатів лабораторних та інструментальних досліджень не були достатні для уточнення діагнозу, дитину було направлено до медико-генетичної консультації НДСЛ «Охматдит», де на підставі даних огляду (клінодактилія пальців стоп, обмеження відведення нижньої щелепи, тугорухливість суглобів), сімейного анамнезу (ураження шийного відділу хребта у батька), даних інструментального обстеження (наявність екзостозів, осифікатів, зрощення хребців, вкорочені та широкі шийки стегнових кісток) встановлений основний діагноз прогресуючої осифікуючої фібродисплазії, запропонована ДНК-діагностика — пошук мутацій гена ACVR-1 для його підтвердження (в роботі). Дитина отримує лікування — диклофенак натрій у протизапальній дозі. З батьками проведена бесіда про перебіг та прогноз захворювання.
Описані випадки дещо відрізняються за особливостями дебюту і перебігу хвороби, швидкістю розвитку осифікуючих процесів у скелетних м’язах. Оскільки темпи прогресування були вищими в першому випадку, дитина одразу розглядалася як пацієнт із м’язовим ураженням, і диференційний діагноз проходив у межах новоутворень, запальних та незапальних уражень м’язів, інших причин їх кальцифікації. У другому випадку на перший план виходить супутнє ураження суглобів, тому діагностичний пошук спочатку був спрямований на уточнення характеру суглобового синдрому, причому наявністю «екзостозів» та інших змін з боку опорно-рухового апарату нехтували. На це впливали й особливості розвитку дитячої ревматології в нашій країні, які сьогодні іноді супроводжуються гіпердіагностикою ювенільного артриту. На наукових заходах доповідаються описи випадків відсутності ефективності їх терапії, коли у пацієнтів через кілька років вперше виявлені вроджені вади розвитку, в тому числі зі зрощенням хребців.
Тому дитячому ревматологу перед прийняттям рішення про призначення лікування дитині необхідно ретельно переглянути критерії діагнозу хвороби та зіставити їх із клінічними проявами захворювання у дитини. У разі ж виявлення додаткових ознак, невідповідності, завжди доцільно розглянути можливість наявності в дитини іншої хвороби, яка супроводжується подібною симптоматикою, перш за все, вроджених аномалій та генетичних захворювань.
Можливості лікування генетичної патології з кожним роком оновлюються і виникають нові, цікаві та оптимістичні повідомлення щодо ефективності нових методів терапії. Незважаючи на те що рідкісні генетичні патології в щоденній практиці лікарів більшості спеціальностей трапляються надзвичайно рідко, вкрай важливо вміти вчасно їх розпізнати та направити цих пацієнтів на подальше обстеження, оскільки від цього залежатимуть перспективи та якість життя не лише наших маленьких пацієнтів, але й їхніх сімей.
Список використаної літератури
- Антелава О.А., Никишина И.П., Гусева И.А. и др. (2015) Прогрессирующая оссифицирующая фибродисплазия. РМЖ, 7: 415–420.
- Гаусманова-Петрусевич И. (1971) Мышечные заболевания. Варшава, с. 326.
- Пономарев А.А., Куликов Е.П., Караваев Н.С., Федосеев А.В. (1998) Редкие кожно-висцеральные синдромы. Рязань, 648 с.
- Проскурова В.И., Костенко И.Н. (1973) О множественном прогрессирующем миозите (оссифицирующем). Врачеб. дело, 4: 114–118.
- Blaszczyk M., Majewski S., Brzezinska-Wcislo L. (2003) Jablonska Fibrodysplasia ossificans progressiva. Eur. J. Dermatol., 13(3): 234–237.
- Clementia Pharmaceuticals Initiates Phase 2 Study of Palovarotene in Patients with Fibrodysplasia Ossificans Progressiva (FOP) (2014) July 14 (http://clementiapharma.com/wp-content/uploads/2019/04/Clementia-20140714.pdf).
- Favus M.J. (1990) Fibrodisplasia (myositis) ossificans progressiva. Primer on metabolic bone diseases and disorders of mineral metabolism. First ed. ASBMR, 297 p.
- Glaser D.L., Economides A.N., Wang L. et al. (2003) In vivo somatic cell gene transfer of an engineered Noggin mutein prevents BMP4–induced heterotopic ossification. Bone Joint. Surg. Am., 85–A(12): 2332–2342.
- Hegyi L., Gannon F.H., Glaser D.L. et al. (2003). Stromal cells of fibrodysplasia ossificans progressiva lesions express smooth muscle lineage markers and the osteogenic transcription factor Runx2/Cbfa-1: clues to a vascular origin of heterotopic ossification? Pathol., 201(1): 141–148.
- Kaplan F.S., Glaser D.L., Shore E.M. et al. (2005) The phenotype of fibrodysplasia ossificans progressiva. Clin. Rev. Bone Miner. Metab., 3: 183–188.
- Lees-Shepard J.B., Yamamoto M., Biswas A.A. et al. (2018) Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nature communic., 9(1): 471.
- Lucotte G., Bathelier C., Mercier G. et al.; FOP Consortium. Fibrodysplasia Ossificans Progressiva Consortium (2000) Localization of the gene for fibrodysplasia ossificans progressiva (FOP) to chromosome 17q21–22. Genet Couns., 11(4): 329–334.
- Mahboubi S., Glaser D.L., Shore E.M., Kaplan F.S. (2001) Fibrodysplasia ossificans progressiva. Pediatr. Radiol., 31(5): 307–314.
- Olmsted E.A., Kaplan F.S., Shore E.M. (2003) Bone morphogenetic protein-4 regulation in fibrodysplasia ossificans progressiva. Clin. Orthop., 408: 331–343.
- Palhares D.B., Leme L.M. (2001) A perspective on the control of myositis ossificans progressive. J. Pediatr. (Rio. J.), 77(5): 431–434.
- Scarlett R.F., Rocke D.M., Kantanie S. et al. (2004) Influenza-like viral illnesses and flare-ups of fibrodysplasia ossificans progressiva. Clin. Orthop., 423: 275–279.
- Semonin O., Fontaine K., Daviaud C. et al. (2001) Identification of three novel mutations of the noggin gene in patients with fibrodysplasia ossificans progressiva. Am. J. Med. Genet., 102 (4): 314–317.
- www.ifopa.org
Редкие болезни: два случая прогрессирующей оссифицирующей фибродисплазии в практике детского ревматолога
1Национальная медицинская академия последипломного образования им. П.Л. Шупика МЗ Украины, Киев
2ДУ «Институт педиатрии, акушерства и гинекологии им. акад. Е.М. Лукьяновой НАМН Украины», Киев
3НДСБ «Охматдет», Киев
Резюме. Прогрессирующая оссифицирующая фибродисплазия является редким (1 из 2 млн) генетическим заболеванием с аутосомно-доминантным типом наследования, которое заключается в постепенной замене мышечной ткани костной и приводит к инвалидизации, а затем и смерти пациента. Путь к установлению диагноза зачастую занимает несколько лет из-за низкой распространенности патологии и клинической схожести с ортопедическими и онкологическими заболеваниями. На сегодняшний день эта болезнь является неизлечимой, но в мире продолжаются клинические испытания препаратов, которые позволят приостановить процесс оссификации. Цель: привлечь внимание к особенностям течения и трудности диагностики прогрессирующей оссифицирующей фибродисплазии. Объект: пациенты педиатрического профиля с прогрессирующей оссифицирующей фибродисплазией. Методы: обработка данных медицинской документации. Результаты: описаны два случая прогрессирующей оссифицирующей фибродисплазии у детей, находившихся на лечении в клинике ГУ «ИПАГ им. акад. Е.М. Лукьяновой НАМН Украины». Выводы: настороженность детских ревматологов и врачей смежных специальностей к выявлению случаев прогрессирующей оссифицирующей фибродисплазии позволит как можно раньше предоставлять всю возможную помощь пациентам с этим диагнозом и улучшать их качество жизни.
Ключевые слова: прогрессирующая оссифицирующая фибродисплазия, клинический случай, дети, орфанные заболевания.
Адреса для листування:
Ошлянська Олена Анатоліївна
04112, Київ, вул. Дорогожицька, 9
Національна медична академія
післядипломної освіти ім. П.Л. Шупика
Leave a comment