IgG4-ЗАЛЕЖНЕ ЗАХВОРЮВАННЯ: СУЧАСНИЙ СТАН ПРОБЛЕМИ Й ОПИС КЛІНІЧНОГО ВИПАДКУ
Яременко О.Б., Коляденко Д.І., Петелицька Л.Б.
Резюме. У статті наведено огляд даних літератури про клінічні прояви IgG4-залежного захворювання, особливості діагностики та сучасні підходи до лікування. Описано клінічний випадок IgG4-залежного захворювання у пацієнтки з ураженням періорбітальних тканин, слізних і великих слинних залоз.
Вступ
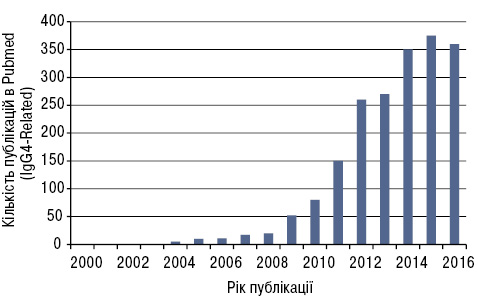
IgG4-залежне захворювання (IgG4-ЗЗ) виділено в окрему нозологічну одиницю лише близько 10 років тому, і з того часу заінтересованість науковців цим патологічним станом стрімко зростає (рис. 1) [3].
IgG4-ЗЗ — фіброзно-запальний стан, що імітує багато злоякісних, інфекційних та запальних захворювань [14]. IgG4-ЗЗ характеризується утворенням склеротичних, пухлиноподібних мас, що містять щільні лімфоплазмоцитарні інфільтрати зі значною кількістю IgG4-плазматичних клітин [23].
Різноманітні прояви IgG4-ЗЗ описані ще у XIX ст. Хвороба Мікуліча (ХМ), тиреоїдит Ріделя та пухлина Кютнера вважалися окремими рідкісними захворюваннями. Першим кроком до відкриття IgG4-ЗЗ був опис у 1995 р. імуноопосередкованої форми панкреатиту, відомого сьогодні як аутоімунний панкреатит (AIП) І типу. Згодом, у 2001 р., H. Hamano та співавтори [6] повідомили про підвищення рівня IgG4 у сироватці крові пацієнтів з АІП і також описали характерну гістологічну структуру при супутньому ретроперитонеальному фіброзі (РПФ), таким чином започаткувавши шлях до визнання системної природи IgG4-ЗЗ [10]. Раніше для опису IgG4-ЗЗ використовували різні терміни: IgG4-пов’язане аутоімунне захворювання, IgG4-пов’язане системне захворювання, IgG4-асоційований мультифокальний системний фіброз, системний IgG4-плазмоцитарний синдром (SIPS), IgG4-пов’язаний мультиорганний лімфопроліферативний синдром (IgG4-MOLPS) [1]. Лише у 2012 р. було прийнято уніфіковану номенклатуру IgG4-ЗЗ. Цього ж року було запропоновано діагностичні критерії IgG4-ЗЗ та розроблено міжнародні консенсусні рекомендації щодо ведення хворих на IgG4-ЗЗ. У 2018 р. на щорічному з’їзді Американського коледжу ревматологів (ACR) затверджено проект класифікаційних критеріїв IgG4-ЗЗ [16].
Епідеміологія
IgG4-ЗЗ вважається рідкісним захворюванням, але його реальну епідеміологію досі не визначено. Доступні епідеміологічні дані базуються переважно на японських популяційних дослідженнях: за даними К. Uchida та співавторів (2012), річна поширеність IgG4-ЗЗ становить 0,28–1,08 випадків на 100 тис. населення [18]. IgG4-ЗЗ зазвичай уражає осіб середнього та старшого віку з дебютом переважно у віці 50–70 років, хоча також описано рідкісні випадки у дітей [10]. Хворіють переважно чоловіки, особливо при залученні панкреатобіліарної системи, де співвідношення чоловіків і жінок становить 3:1. Гендерні відмінності менш виражені у пацієнтів із залученням слинних залоз [21].
Етіологія та патогенез
Етіопатогенез IgG4-ЗЗ залишається невідомим. Т-клітини — найпоширеніші клітини лімфоплазмоцитарного інфільтрату при IgG4-ЗЗ — вважаються чинниками його патогенезу. Розвиток захворювання пов’язують з імунними реакціями за участю як T-хелперів 2 (Th2), так і регуляторних T-клітин (Treg). Th2 продукують інтерлейкін (IЛ)-4, IЛ-5, ІЛ-10 та ІЛ-13 і регулюють синтез IgG4 та IgE. Натомість Treg продукують IЛ-10 і трансформуючий фактор росту β (TGF-β), стимулятор активності фібробластів). Вважають, що Th2 стимулюються шляхом постійної антигенної презентації В-клітинами та плазмобластами. Наслідками повторних циклів антигенної стимуляції та секреції цитокінів є фіброз тканин та лімфоплазмоцитарна інфільтрація зі значною кількістю IgG4-плазматичних клітин [23]. Проте нещодавні дані ставлять під сумнів провідну роль Т-лімфоцитів у патогенезі IgG4-ЗЗ. Зокрема, доведено, що при IgG4-ЗЗ гладкі клітини продукують значну кількість IЛ-4, IЛ-10 і TGF-β. Останні дослідження виокремлюють роль інших цитокінів, зокрема IЛ-21 та IЛ-27, що продукуються макрофагами та іншими антигенпрезентуючими клітинами і беруть участь у диференціації Т-хелперів. Крім того, макрофаги M2, активовані цитокінами Т-хелперів і Т-регуляторів (ІЛ-4, ІЛ-13, ІЛ-18), імовірно, теж беруть участь у різних процесах фіброзу. Toll-подібні рецептори та домени нуклеотидзв’язувальної олігомеризації доменів макрофагів стимулюють подальшу активацію та проліферацію IgG4-В-клітин. Зростає інтерес також до ролі базофілів та еозинофілів при IgG4-ЗЗ. Отже, значна активація вродженого імунітету може свідчити про наявність імунної відповіді проти ще невідомого тригерного антигену [9].
Серед генетичних чинників розглядають серотипи HLA DRB1*0405 i DQB1*0401 та однонуклеотидні поліморфізми у деяких не-HLA-генах (FCRL3 i CTLA4) [2]. Деякі дослідження показали можливу роль молекулярної мімікрії за участю Helicobacter pylori. Описано імунокомплексні депозити в підшлунковій залозі, нирках, а також наявність аутоантитіл до лактоферину і карбоангідрази ІІ [12].
Результати останніх досліджень свідчать про підвищення рівнів CD19CD20CD27CD38 плазмобластів у периферичній крові, що корелюють з активністю захворювання. Оскільки цим клітинам притаманна олігоклональна експансія та соматична гіпермутація, можна припустити, що при IgG4-ЗЗ наявна специфічна антигенна імунна відповідь. Рецидиви IgG4-ЗЗ після терапії ритуксимабом (RTX) асоціюються з появою нових ліній плазмобластів, що, ймовірно, свідчить про залучення наївних В-клітин з червоного кісткового мозку [10].
IgG4 — найменш численний підклас IgG, що становить менше 5% загального IgG (норми його рівня у сироватці крові коливаються від 0,05 до 1,4 г/л) і вважається незапальним імуноглобуліном через обмежену здатність фіксувати комплемент та зв’язувати Fc-рецептори [11]. Крім того, завдяки нестабільним зв’язкам міжвузлових ланцюгів, IgG4 може обмінюватися своїми Fab-фрагментами з іншими молекулами IgG4 (рис. 2), що робить їх двовалентними антитілами із двома різними антигензв’язувальними фрагментами, а це унеможливлює утворення імунних комплексів [7].
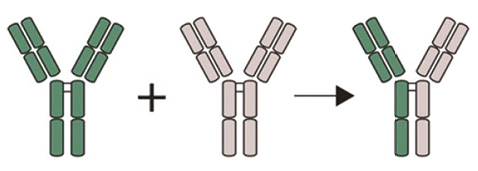
IgG4 відіграє важливу роль у таких імуноопосередкованих станах, як вульгарна пухирчатка (IgG4-антитіла до десмоглеїну відповідають за формування шкірних пухирців), ідіопатичний мембранозний гломерулонефрит та ідіопатична тромбоцитопенічна пурпура [15]. Проте у 20–40% пацієнтів з IgG4-ЗЗ рівень IgG4 у сироватці крові може бути в нормальних діапазонах. З іншого боку, підвищений рівень IgG4 можливий при різних захворюваннях: синдром Шегрена (СШ — 7%), системний червоний вовчак (СЧВ — 10%), ревматоїдний артрит (за даними Y. Chen та співавторів (2014), у 46% пацієнтів з ревматоїдним артритом спостерігався підвищений рівень IgG4 у сироватці крові, що корелював з активністю захворювання [4]), рак, хвороба Кастлемана, алергічні захворювання, еозинофільний гранулематоз із поліангіїтом, саркоїдоз, а також у здоровій популяції (2%) [13].
Патоморфологія
Ключовими морфологічними ознаками IgG4-ЗЗ є: 1) щільний лімфоплазмоцитарний інфільтрат з високим вмістом IgG4-плазматичних клітин; 2) спіралевидний фіброз; 3) флебіт з облітерацією просвіту судин. Інші гістопатологічні зміни представлені флебітом без облітерації просвіту судин і підвищеним вмістом еозинофілів. Важливе значення має імуногістохімічне дослідження з кількісним визначенням IgG4-плазматичних клітин. Для IgG4-ЗЗ характерна наявність ≥10 IgG4-плазматичних клітин і/або співвідношення IgG4-експресуючих плазматичних клітин до IgG-експресуючих плазматичних клітин (індекс IgG4/IgG) >40% [1].
Класифікація
Досі немає загальноприйнятої класифікації IgG4-ЗЗ. Перелік нозологічних одиниць щорічно переглядається та доповнюється (табл. 1).
| Тканина, орган | Класичний термін | IgG4-пов’язаний термін |
|---|---|---|
| Підшлункова залоза | АІП | IgG4-пов’язаний панкреатит (АІП І типу) |
| Жовчні протоки, жовчний міхур і печінка | Склерозуючий холангіт | IgG4-пов’язаний склерозуючий холангіт
IgG4-пов’язаний холецистит IgG4-пов’язана гепатопатія |
| Щитоподібна залоза | Тиреоїдит Ріделя
Тиреоїдит Хашімото |
IgG4-пов’язане захворювання щитоподібної залози |
| Слинні і слізні залози | ХМПухлина Кютнера | IgG4-пов’язаний дакріоаденіт
IgG4-пов’язаний сіалоаденіт IgG4-пов’язаний паротит IgG4-пов’язане захворювання підщелепних залоз |
| Орбіта | Пухлина орбіти | IgG4-пов’язане офтальмологічне захворювання
IgG4-пов’язана запальна псевдопухлина орбіти IgG4-пов’язане панорбітальне запалення IgG4-пов’язаний орбітальний міозит |
| Легені, середостіння, плевра | Інтерстиційна пневмонія | IgG4-пов’язане захворювання легень
IgG4-пов’язаний медіастиніт IgG4-пов’язаний плеврит |
| Нирки | Тубулоінтерстиційний нефрит (ТІН) | IgG4-пов’язане захворювання нирок
ТІН у межах IgG4-ЗЗ Мембранозний гломерулонефрит у межах IgG4-ЗЗ |
| Заочеревинний простір, артерії | РПФ (хвороба Ормонда)
Лімфоплазмоцитарний аортит Запальна аневризма аорти |
IgG4-пов’язаний РПФ
IgG4-пов’язаний аортит/періаортит IgG4-пов’язаний періартеріїт |
| Шкіра | Псевдолімфома шкіри | IgG4-пов’язане захворювання шкіри |
| Гіпофіз | Аутоімунний гіпофізит | IgG4-пов’язаний гіпофізит |
| Мозкові оболонки | Гіпертрофічний пахіменінгіт | IgG4-пов’язаний пахіменінгіт |
| Лімфатичні вузли | IgG4-пов’язана лімфаденопатія | |
| Молочна залоза | IgG4-пов’язаний мастит | |
| Передміхурова залоза | IgG4-пов’язаний простатит | |
| Інші | IgG4-пов’язане периневральне захворювання
IgG4-пов’язаний мезентерит IgG4-пов’язаний епідидимоорхіт IgG4-пов’язана паратестикулярна псевдопухлина IgG4-пов’язаний перикардит |
Клініка
Початок IgG4-ЗЗ зазвичай є підгострим. У пацієнтів часто відсутня будь-яка симптоматика, і захворювання діагностується випадково під час обстеження з приводу інших причин [13]. Можлива також спонтанна ремісія з багаторічною неактивністю захворювання [14]. Важливою ознакою захворювання є позитивна клінічна відповідь на терапію ex juvantibus глюкокортикоїдами (ГК) [2].
Хвороба була зареєстрована практично в кожній системі органів [14]. Приблизно у 40% пацієнтів захворювання дебютує з ураження одного органа у формі об’ємного утворення, яке викликає специфічні прояви залежно від локалізації. Симптоми варіюють від набряку уражених органів (слинних та слізних залоз, лімфатичних вузлів) до обструкції (проток підшлункової залози, сечоводів), дисфункції органів (гіпофізарна недостатність внаслідок гіпофізиту, ниркова недостатність), і навіть невідкладних станів (гострий аортальний синдром, пахіменінгіт, панкреатит). У невеликої кількості пацієнтів відзначають конституційні симптоми, такі як лихоманка та втрата маси тіла [23]. Найтиповіші клінічні прояви наведено у табл. 2 [4].
| Клінічні прояви | Частка пацієнтів із цим симптомом, % |
|---|---|
| Набряк підщелепних залоз | 51,0 |
| Набряк слізних залоз | 42,0 |
| Збільшення поверхневих лімфовузлів | 37,0 |
| Біль у животі | 35,0 |
| Набряк привушних залоз | 24,0 |
| Закладеність носа | 21,5 |
| Жовтяниця | 14,0 |
| Свербіж | 13,5 |
| Кашель | 13,5 |
| Біль у нижній ділянці спини | 13,5 |
| Розлади сечовипускання | 13,0 |
| Ксерофтальмія/ксеростомія | 12,5 |
| Нудота і/та блювання | 12,0 |
| Лихоманка | 9,0 |
| Набряки | 8,5 |
| Екзофтальм | 5,5 |
| Артралгія | 4,0 |
| Збільшення щитоподібної залози | 2,5 |
| Алергологічний анамнез | 59,0 |
| Відсутність симптомів | 1,0 |
Також проаналізовано варіанти поєднання уражених органів і систем (рис. 3). Серед хворих із залученням одного органа 13% становили пацієнти із ХМ, 7,5% — з РПФ/медіастинальним фіброзом (МФ), 7% — з ураженням гепатопанкреатобіліарної системи. Серед осіб із залученням двох органів ХМ діагностували найчастіше (26,5%). Крім того, комбінація ХМ із залученням двох інших органів становила 14,0% випадків, тоді як пацієнти з ураженням трьох органів без ХМ становили лише 1,5% випадків. Окрім того, частка пацієнтів із множинним залученням органів (≥3) становила 12,0%, у більшості з них також виявлено ХМ. Таким чином, оцінено рівень впевненості (сильний, помірний чи слабкий) щодо індикації IgG4-ЗЗ відповідно до поєднання уражених органів (табл. 3) [4].
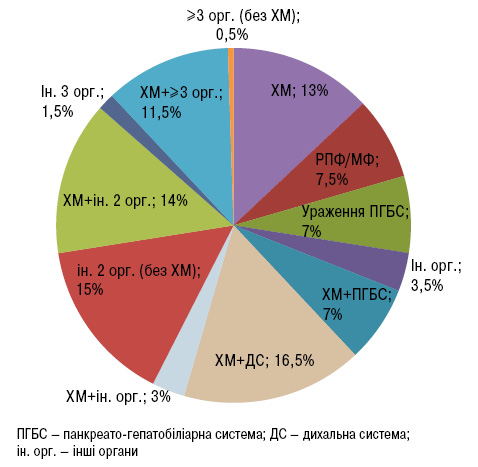
до типу залученого органа
| Тип ураженого органа | Рівень впевненості щодо індикації IgG4-ЗЗ |
|---|---|
| Залучення одного типу органа | |
| Слинні залози/слізні залози (ХМ) | Сильний |
| Панкреатогепатобіліарна система | Помірний |
| РПФ/МФ | Помірний |
| Регіонарна/системна лімфаденопатія | Слабкий |
| Дихальна система (легені/дихальні шляхи/плевра) | Слабкий |
| Сечостатева система (нирки/сечоводи/сечовий міхур/передміхурова залоза) | Слабкий |
| Залучення двох типів органів | |
| ХМ + дихальна система | Сильний |
| ХМ + панкреатогепатобіліарна система | Сильний |
| ХМ + інший тип органа | Помірний |
| РПФ/МФ + панкреатогепатобіліарна система | Помірний |
| 2 інших типи органів | Слабкий |
| Залучення трьох типів органів | |
| ХМ + панкреатогепатобіліарна + дихальна система | Сильний |
| ХМ + пакреатогепатобіліарна система + РПФ/МФ | Сильний |
| ХМ + пакреатогепатобіліарна система + сечостатева система | Помірний |
| ХМ + 2 інших типи органів | Помірний |
| 3 інші типи органів | Слабкий |
| Множинне ураження органів | |
| ХМ + ≥3 типів органів | Сильний |
| ≥3 типів органів без ХМ | Помірний |
Голова та шия
Ураження слізних залоз та м’яких тканин орбіти часто може бути першим проявом захворювання. Типовими симптомами є одно- чи двобічний набряк м’яких тканин орбіти без порушення гостроти зору і симптомів сухого кератокон’юнктивіту. При ураженні м’язів очного яблука можливе виникнення порушення їх функції, а при компресії очного нерва — зорові розлади. Особливо характерним є залучення слинної підщелепної залози, що носить назву пухлини Кютнера. При одночасному симетричному ураженні слізних і слинних залоз патологія має назву ХМ [1]. Важливим є проведення диференційної діагностики між ХМ та СШ. ХМ має ряд відмінностей від класичного СШ, що полягають у: 1) різниці гендерного розподілу (ХМ виникає з однаковою частотою як у чоловіків, так і у жінок, тоді як СШ — переважно у жінок); 2) стійкому збільшенні слізних і слинних залоз; 3) нормальній секреції чи незначному порушенні секреції залоз; 4) відповіді на терапію ГК; 5) гіпергаммаглобулінемії (за рахунок IgG4), підвищенні рівня IgЕ і низькій частоті виявлення антинуклеарних, анти-SS-A- і SS-B-антитіл та ревматоїдного фактора в серологічних аналізах; 6) частому поєднанні з розвитком АІП І типу; 7) формуванні кількох гермінативних центрів у залозистій тканині. СШ характеризується перидуктальною лімфоцитарною інфільтрацією з атрофією чи значним руйнуванням ацинусів (малюнок «дерева» за даними сіалографії), а при ХМ виявляється позаперидуктальна лімфоцитарна інфільтрація з гіперпластичними гермінативними центрами, помірне руйнування ацинусів без ураження проток. У пацієнтів із СШ, на відміну від ХМ, ніколи не виникає ізольоване ураження підщелепної слинної залози [2].
Прояви з боку ЛОР-органів включають алергічний риніт, поліпи носа, хронічний синусит і фарингіт. Об’ємні утворення можуть виникати в приносових пазухах, середньому вусі та кістках лицевого черепа. Описано випадки трахеїту й ураження голосових зв’язок. Збільшення щитоподібної залози внаслідок тиреоїдиту Ріделя може супроводжуватися болем у ділянці шиї, задишкою, дисфагією та дисфонією [14]. У ⅔ хворих виявляють антитиреоїдні антитіла. Часто до патологічного процесу залучаються також паращитоподібні залози з розвитком симптомів гіпопаратиреозу [2].
Підшлункова залоза
АІП І типу вважається панкреатичним проявом IgG4-ЗЗ і характеризується незначно вираженими абдомінальними симптомами, зазвичай без гострих атак панкреатиту, дифузним збільшенням підшлункової залози, звуженням панкреатичної протоки та безболісною обструктивною жовтяницею, що нерідко симулює аденокарциному підшлункової залози [1]. У ретроспективному дослідженні Інституту Джона Хопкінса відзначено, що у 2,5% хворих, які перенесли панкреатодуоденектомію у зв’язку з раком підшлункової залози, гістологічно діагностовано AIП І типу [2].
Біліарний тракт і печінка
IgG4-подібний склерозуючий холангіт у 50–90% пацієнтів поєднується з АІП І типу [1]. За відсутності лікування він може прогресувати до термінальної стадії захворювання печінки. Наявність об’ємних утворень часто супроводжується механічною жовтяницею [14].
Легені
Ураження дихальної системи виникає у 14–50% пацієнтів із IgG4-ЗЗ. У половини хворих ураження легень має симптоматичний перебіг (кашель, задишка, біль у грудній клітці, кровохаркання). Спектр клінічних проявів включає псевдопухлину, інтерстиційну пневмонію, медіастинальну лімфаденопатію, МФ, стеноз дихальних шляхів та плеврит [2]. Описано 4 основні рентгенологічні патерни IgG4-пов’язаного захворювання легень: 1) щільні вузлики; 2) бронховаскулярний патерн (з потовщенням бронхосудинних пучків і міжчасткових перегородок); 3) альвеолярно-інтерстиційний патерн (бронхоектази, дифузні затемнення типу «матового скла»); 4) округлі затемнення типу «матового скла» [12].
Лімфатичні вузли
Лімфаденопатія виявляється у 80% хворих з АІП і є загальним симптомом IgG4-ЗЗ. Зазвичай до патологічного процесу залучається кілька груп лімфатичних вузлів (найчастіше лімфатичні вузли середостіння, внутрішньочеревні й пахвові). Лімфатичні вузли, як правило, невеликі, безболісні, конституційні симптоми (лихоманка і втрата маси тіла) відсутні, а рівень лактатдегідрогенази нормальний чи мінімально підвищений [2].
Нервова система
Залучення нервової системи — дуже рідкісне, найчастіше уражається гіпофіз (гіпопітуїтаризм, нецукровий діабет та локальне об’ємне утворення), мозкові оболонки (гіпертрофічний пахіменінгіт) та периферичні нерви. Особливістю ураження периферичної нервової системи є відсутність неврологічної симптоматики за рахунок інфільтрації епіневрію та цілісності нервового пучка (периневральне захворювання) [2]. IgG4-ЗЗ зазвичай не вражає паренхіму головного мозку [14].
Заочеревинний простір, нирки, великі судини
РПФ — хронічне запальне захворювання з вираженим фіброзом тканин заочеревинного простору. РПФ здебільшого виникає у чоловіків середнього віку й асоціюється з тютюнопалінням. Клінічно у хворих із двобічним масивним розростанням щільної волокнистої сполучної тканини в ретроперитонеальній клітковині виникає двобічна обструкція сечоводів з розвитком больового синдрому та анурії. Найпоширенішими ураженнями нирок при IgG4-ЗЗ є тубулоінтерстиційний нефрит і мембранозний гломерулонефрит. Клінічна картина зазвичай невиразна, хоча можуть бути наявними нефротичний синдром і хронічна ниркова недостатність. Захворювання нирок супроводжується досить низьким рівнем комплементу. IgG4-залежний аортит зазвичай має безсимптомний перебіг і може дебютувати розривом аневризми аорти. Характерною ознакою IgG4-залежного аортиту за даними контрастної комп’ютерної томографії (КТ) є кругове потовщення артеріальної стінки, що спричинене запаленням і склерозом адвентиції та накопичення 18-фтордезоксиглюкози за даними позитронно-емісійної томографії (ПЕТ) [2].
Інші органи
До 40% пацієнтів із IgG4-ЗЗ мають такі алергічні прояви, як атопія, екзема, бронхіальна астма, хронічний синусит [13]. Шкірні прояви включають еритематозні бляшки та підшкірні вузлики, які мають тенденцію до локалізації на обличчі або голові і супроводжуються виникненням свербежу (шкірна псевдолімфома). IgG4-асоційований мастит може проявлятися у вигляді одного або декількох безболісних утворень на одній або обох молочних залозах, з/без ознак системного процесу. У чоловіків IgG4-ЗЗ може бути причиною простатиту, орхіту, а також паратестикулярної псевдопухлини. Ще одним проявом цього захворювання є ураження перикарда, яке проявляється симптомами констриктивного перикардиту [2].
Лабораторна діагностика
Немає стандартного лабораторного показника, який би чітко підтвердив IgG4-ЗЗ. Зміни маркерів запального процесу, таких як швидкість осідання еритроцитів і С-реактивний білок, не є характерними для цього захворювання і не корелюють з його активністю. Антинуклеарні антитіла і ревматоїдний фактор виявляють у низьких титрах у 30 та 20% пацієнтів із IgG4-ЗЗ. Виявлення інших більш специфічних аутоантитіл, таких як антитіла до SS-A та SS-B, до двоспіральної ДНК, антинейтрофільні антитіла не є характерною ознакою для цієї патології та потребує виключення інших ревматичних захворювань (СШ, СЧВ, системні васкуліти) [2]. Часто спостерігається низький рівень комплементу, особливо при залученні нирок. Також можливі зміни у біохімічному аналізі крові, що свідчить про порушення функції нирок, підшлункової залози та печінки (підвищення аланінамінотрансферази, аспартатамінотрансферази, гамма-глютамілтранспептидази, креатиніну). Крім того, у 40% пацієнтів наявна периферична еозинофілія та підвищення рівня IgE у сироватці крові [10]. Підвищення рівня IgG4 у сироватці крові донедавна відігравало основну роль у встановленні діагнозу IgG4-ЗЗ, але дані останніх років свідчать про його низьку діагностичну цінність. Так, у 30% пацієнтів із IgG4-ЗЗ визначаються нормальні рівні IgG4 в сироватці крові. Z.S. Wallace та співавтори (2015) [22] припускають перспективну роль проточної цитометрії, а саме визначення рівня циркулюючих плазмобластів (CD19lowCD38+CD20-CD27+) у діагностиці та оцінці перебігу IgG4-ЗЗ. За даними моноцентрового дослідження підвищення рівня циркулюючих плазмобластів виявляється навіть у хворих з нормальним рівнем IgG4, а їх рівень корелює з активністю захворювання [2].
Інструментальна діагностика
Основним методом інструментального обстеження хворих на IgG4-ЗЗ є КТ та магнітно-резонансна томографія, які можна використовувати для оцінки поширення процесу та контролю за перебігом захворювання, проте часто не дають змоги відрізнити IgG4-ЗЗ від злоякісних новоутворень. Корисною є також ПЕТ із 18-фтордезоксиглюкозою, що дозволяє виявити активні запальні ураження і, таким чином, оцінити ступінь поширеності захворювання та реакцію на лікування, а також визначити точне місце для біопсії [10]. Широко використовують також холангіопанкреатографію та ультразвукове дослідження.
Діагностика
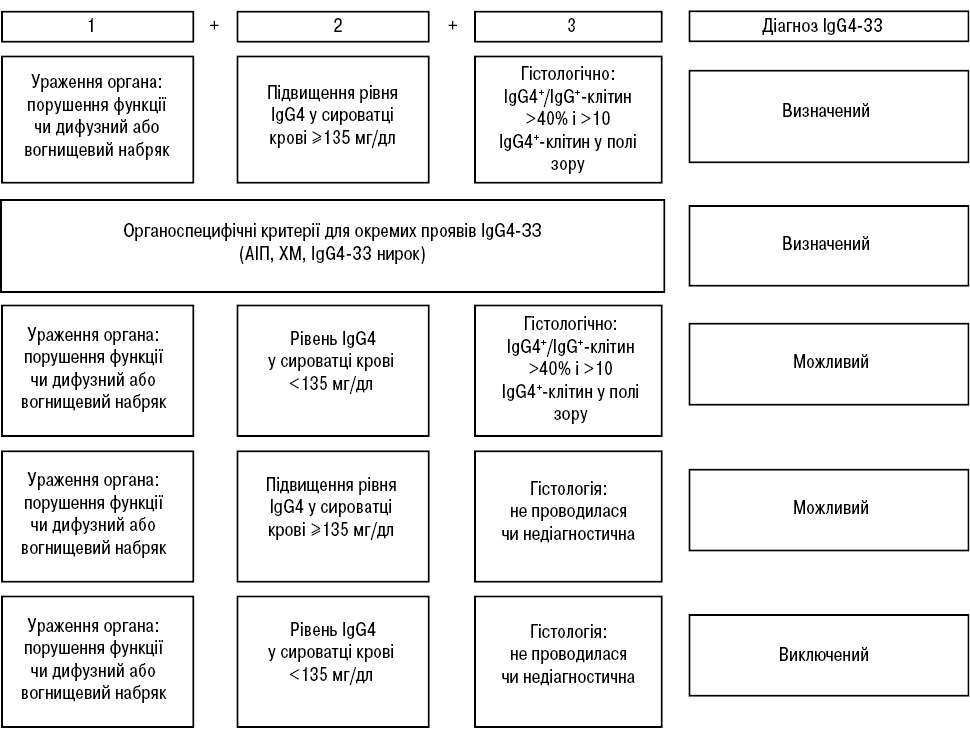
Для визначення IgG4-ЗЗ використовують діагностичні критерії H. Umehara та співавторів (2017) [19], що базуються на клінічних, імунологічних і гістопатологічних ознаках (табл. 4, рис. 4).
| Дослідження | Характерні ознаки | Діагноз IgG4-ЗЗ |
|---|---|---|
| Клінічне (1) | Дифузне/локальне збільшення чи пухлиноподібне утворення у одному чи кількох органах | Достовірний: 1 + 2 + 3 |
| Імунологічне (2) | Підвищення вмісту IgG4 у сироватці крові (>135 мг/дл) | Імовірний: 1 + 3 |
| Гістологічне (3) | Виражена лімфоцитарна і плазмоцитарна інфільтрація і фіброз.Інфільтрація IgG4-плазматичними клітинами (індекс IgG4/IgG4 >40% і >10 IgG4-плазматичних клітин у полі зору) | Можливий: 1 + 2 |
У 2018 р. на щорічному конгресі ACR у Чикаго (США) затверджено проект класифікаційних критеріїв IgG4-ЗЗ (специфічність — 99,2%, чутливість — 85,5%) [16]. Проект включає критерії включення та виключення. Першим кроком у встановленні діагнозу IgG4-ЗЗ є ідентифікація ураження принаймні одного органа з переліку 10 органів, залучення яких є типовим для захворювання: підшлункова залоза, жовчні протоки, орбіти, слізні залози, великі слинні залози, заочеревинний простір, нирки, аорта, м’які оболонки мозку, щитоподібна залоза. У пацієнтів без залучення принаймні одного з цих органів виключається імовірність IgG4-ЗЗ. Наступним кроком є виявлення пацієнтів з наявним принаймні одним із критеріїв виключення (табл. 5). Завершальним етапом є виявлення достатньої кількості класифікаційних ознак для остаточного підтвердження IgG4-ЗЗ (табл. 6).
| Клінічні | Лихоманка |
| Відсутність відповіді на терапію ГК | |
| Лейкопенія і тромбоцитопенія | |
| Периферична еозинофілія (>3000/мм) | |
| Серологічні | Позитивність за антитілами до протеїнази-3 або мієлопероксидази |
| Позитивність за антитілами до SS-A чи SS-B | |
| Позитивність за антитілами до двоспіральної ДНК, рибонуклеопротеїну чи антигену Сміта (антитіла до екстрагованих ядерних антигенів) | |
| Кріоглобулінемія | |
| Наявність інших хворобоспецифічних антитіл | |
| Рентгенологічні | Швидка рентгенологічна прогресія |
| Аномалії довгих кісток (наприклад хвороба Ердгейма – Честера) | |
| Спленомегалія | |
| Підозра на інфекцію, злоякісне новоутворення чи їх поєднання | |
| Гістологічні | Первинне гранулематозне запалення |
| Некротизивний васкуліт | |
| Інфільтрація злоякісними клітинами | |
| Виражена гістіоцитарна інфільтрація | |
| Виражена нейтрофільна інфільтрація | |
| Мультицентрична хвороба Кастлемана | |
| Виражений некроз | |
| Ознаки запальної псевдопухлини |
|
Домен |
Бали* |
|
|
Рівень IgG4 |
У нормі Вище норми у <2 разів Вище норми у 2–5 разів Вище норми у >5 разів |
0 3,7 6,1 10,8 |
|
Гістологічне та імуногістохімічне дослідження |
Неінформативна біопсія Щільні лімфоплазмоцитарні інфільтрати (ЩЛІ) ЩЛІ + облітеруючий флебіт ЩЛІ + спіралевидний фіброз |
0 3,7 6,1 13,3 |
|
Збільшення слізних та великих слинних залоз |
Ураження однієї групи залоз Ураження двох і більше груп залоз |
5,9 13,8 |
|
Грудна клітка і грудна аорта |
Перибронховаскулярне та септальне потовщення Паравертебральні м’якотканинні пучки у грудній клітці |
3,8 9,8 |
|
Підшлункова залоза і біліарний тракт |
Дифузне збільшення підшлункової залози (втрата часточкової будови) Дифузне збільшення підшлункової залози і капсулоподібний обідок зі зниженою щільністю Залучення підшлункової залози і біліарного тракту |
8,0 10,5 18,7 |
|
Нирки |
Гіпокомплементемія Потовщення ниркової миски або м’яких тканин чи їх поєднання |
5,8 8,1 |
|
Заочеревинний простір |
Дифузне потовщення стінки черевної аорти М’якотканинні структури навколо клубових артерій або аорти нижче відходження ниркових артерій |
4,1 7,8 |
*Для встановлення діагнозу IgG4-ЗЗ необхідно щонайменше 19 балів.
Диференційна діагностика IgG4-ЗЗ потребує виключення наявності злоякісних пухлин і схожих нозологій (табл. 7).
|
Особливо складною може бути диференційна діагностика при дебюті захворювання з очних проявів, оскільки перебіг значної кількості рідкісних захворювань може починатися із залученням орбіт. Зокрема, за даними V. Vasilyev та співавторів (2017), причиною офтальмологічних проявів у 35% пацієнтів є IgG4-ЗЗ, у 29% — гранулематозні захворювання, у 17,5% — неходжкінські лімфоми (табл. 8) [20].
| Непухлинні захворювання | Кількість пацієнтів | % пацієнтів | Гематологічні захворювання | Кількість пацієнтів | % пацієнтів |
|---|---|---|---|---|---|
| 108 | 78,5 | 30 | 21,5 | ||
| IgG4-ЗЗ | 48 | 35,0 | Неходжкінські лімфоми | 20 | 14,5 |
| Гранулематозні хвороби (саркоїдоз, гранулематоз з поліангіїтом) | 41 | 29 | Хвороба Ердгейма – Честера | 1 | 0,7 |
| Аутоімунний дакріоаденіт (недиференційований) | 7 | 5 | AL-амілоїдоз | 3 | 2,1 |
| Ідіопатичне запалення орбіти | 7 | 5 | NK-/T-клітинна лімфома носа | 1 | 0,7 |
| Ендокринна офтальмопатія | 2 | 1,4 | Гістіоцитоз | 4 | 2,9 |
| Синдром Когана | 1 | 0,7 | Ювенільна апоневротична фіброма | 1 | 0,7 |
| Рецидивний поліхондрит | 1 | 0,7 |
У ретроспективному дослідженні C.A. Tiegs-Heiden та співавторів (2014) проаналізовано спектр періорбітальних уражень при IgG4-ЗЗ очей і виявлено, що збільшення зовнішніх м’язів очного яблука (ЗМОЯ) мало місце у 89% пацієнтів, до того ж у 88% воно було двобічним, і у 71% — найбільш залученими були латеральні прямі м’язи. При цьому у 96% пацієнтів не спостерігалось супутнього ураження сухожиль окорухових м’язів. У 70% пацієнтів мало місце залучення слізних залоз, 44% — орбітальної клітковини, 30% — підочного нерва, 89% — приносових пазух (у вигляді потовщення слизової оболонки). Відповідно до цих характеристик було розроблено алгоритм діагностичного пошуку: у пацієнтів з вип’ячуванням очних яблук чи періорбітальним набряком при ураженні слізних залоз та ЗМОЯ (особливо латеральних прямих) за відсутності залучення їх сухожиль, що супроводжується потовщенням слизової оболонки приносових пазух, у першу чергу слід розглядати IgG4-ЗЗ (рис. 5) [17].
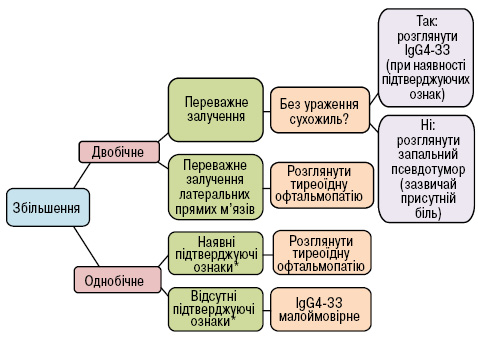
*Підтверджуючі ознаки: ураження слізних залоз, приносових пазух, підочного нерва, орбітальної клітковини.
Тактика лікування
Навіть субклінічне ураження нирок, жовчних проток і підшлункової залози може призвести до тяжких незворотних ушкоджень протягом кількох місяців, тому потребує негайного лікування. Проте неодноразово повідомлялося про спонтанні ремісії та відсутність прогресування захворювання у нелікованих пацієнтів. Отже, вичікувальна тактика може бути доцільною у осіб з незначними симптомами та без ознак дисфункції органів (наприклад при безсимптомній лімфаденопатії та незначному збільшенні слинних залоз) [10]. Показаннями до проведення ургентного лікування IgG4-ЗЗ є наявність аортиту, РПФ, стриктур проксимальних жовчовивідних шляхів, тубулоінтерстиційний нефрит, перикардит, пахіменінгіт і збільшення підшлункової залози [2]. У деяких випадках виникає потреба в терміновому хірургічному втручанні, зокрема при гострому аортальному синдромі, обструкції проток підшлункової залози чи сечоводів.
Початкова та підтримувальна терапія
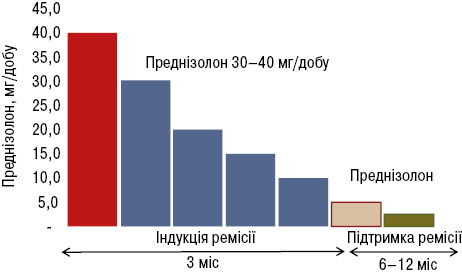
ГК визнано препаратами першої лінії при IgG4-ЗЗ з моменту першого опису AIП у 1995 р., коли помітили швидку відповідь на лікування ГК. Згідно з нещодавно опублікованим консенсусним твердженням щодо лікування IgG4-ЗЗ, переважна більшість експертів все ще вважають ГК препаратами першої лінії для індукції ремісії у всіх хворих з активним IgG4-ЗЗ за відсутності протипоказань (рис. 6). Здебільшого для стартової терапії застосовують преднізолон у дозі 30–40 мг/добу (0,6 мг/кг маси тіла на добу), яку змінюють залежно від активності та тривалості захворювання. Відповідь на терапію ГК зазвичай спостерігається протягом декількох днів або тижнів, і протягом кількох місяців більшість пацієнтів досягають ремісії. Повільне зниження дози ГК необхідно починати через 2–4 тиж після початку терапії (по 10 мг кожні 2 тиж до досягнення дози 20 мг, а потім — по 5 мг кожні 2 тиж). Тривалість підтримувальної терапії низькими дозами ГК (2,5–5 мг/добу преднізолону) на період до 3 років, як це пропонується японськими рекомендаціями щодо AIП, досі залишається об’єктом обговорення. Тривалу підтримувальну терапію слід розглядати при рецидивному перебігу та мультиорганних ураженнях, особливо у разі залучення життєво важливих органів [10].
Експертні думки залишаються невизначеними щодо питання, чи слід застосовувати імуносупресивні засоби у індукційній та підтримувальній терапії, окрім ГК. Згідно з консенсусними рекомендаціями, застосування імуносупресорів доцільне у випадках, коли доза ГК не може бути знижена у зв’язку з постійно високою активністю захворювання. Серед цитостатиків для лікування IgG4-ЗЗ застосовують азатіоприн, мікофенолату мофетил, метотрексат, 6-меркаптопурин, такролімус та циклофосфамід. Проте ефективність цих препаратів не оцінено в проспективних дослідженнях [2].
Останнім часом застосування RTX надало новий імпульс у лікуванні пацієнтів із IgG4-ЗЗ. За даними M. Ebbo та співавторів (2017) [5], клінічну відповідь отримано у 93,5% пацієнтів через 1 міс після початку терапії RTX, навіть у разі стероїдорезистентності. Наприкінці дослідження відміни ГК досягнуто у 51,5% пацієнтів; середня підтримувальна доза ГК становила 9,6±9,3 мг/добу. Проте протягом середнього періоду спостереження (24,8±21 міс) у 41,9% пацієнтів відбувся рецидив захворювання через 19±11 міс після припинення терапії RTX. При цьому спостерігалася достовірна асоціація (р=0,04) між активністю захворювання (IgG4-ЗЗ Responder Index >9) та частотою рецидиву. Натомість підтримувальна терапія RTX достовірно сприяла тривалішому безрецидивному періоду (р=0,02), але водночас супроводжувалася частішими випадками тяжких інфекцій і гіпогаммаглобулінемії [5]. І хоча результати останніх досліджень є перспективними, досі відсутній консенсус щодо дозування та частоти застосування RTX.
Смертність
У нещодавньому систематичному огляді P. Brito-Zeron та співавторів проаналізовано смертність за даними 7 досліджень (294 пацієнти, період спостереження — 29,2 міс) [24]. Летальний кінець зареєстровано у 26 (8,8%) пацієнтів. Причинами смерті були легеневі захворювання, розрив аневризми аорти, холангіт, ниркова недостатність, рак, серцево-судинні та інфекційні захворювання.
Наше спостереження
Пацієнтка М., 24 роки (1993 р.н.), хворіє з 2011 р., коли з’явилася припухлість у лівій підщелепній ділянці. Було проведено КТ щелепно-лицевої ділянки з внутрішньовенним контрастуванням: симетричне збільшення слізних та великих слинних залоз (особливо лівої підщелепної) з гомогенним підвищенням щільності; зниження прозорості усіх синусів і значне потовщення їх слизової оболонки; інші тканини не змінені. У загальному аналізі крові та сечі — без змін. Антитіла до двоспіральної ДНК (анти-dsDNA), Ro(SS-A), La(SS-B), топоізомерази-1 (анти-Scl-70) — негативні. Остаточний діагноз встановлено не було. Хворій проведено хірургічне лікування: видалено частину лівої слинної піднижньощелепної залози (гістологічно — неспецифічна гіперплазія залози), але вже через кілька місяців знову з’явилася припухлість цієї ж ділянки. Ще через деякий час приєдналися офтальмологічні прояви — набряк над очними яблуками, більше зліва, потім екзофтальм, набряк під очима; з’явилися шийна лімфаденопатія та припухлість привушних ділянок. У грудні 2012 р. проведено біопсію трьох структур із такими результатами гістологічного дослідження: 1) ліва підщелепна залоза: значна лімфоплазмоцитарна інфільтрація, склероз строми, що відповідає хронічному склерозуючому сіалоаденіту. Імуногістохімічне дослідження: лімфоцитарні інфільтрати складаються з Т- і В-клітин; значна кількість IgG4-позитивних плазмоцитів (>75 у полі зору), індекс IgG4/IgG — приблизно 80%; 2) привушна залоза: ознаки лімфоцитарного сіалоаденіту; 3) лімфатичний вузол із задньої поверхні шиї: ознаки реактивної фолікулярної гіперплазії.
За даними клінічної картини (дифузне збільшення слинних і слізних залоз, лімфаденопатія) та гістологічного дослідження (індекс IgG4/IgG4 >40% і >10 IgG4-плазматичних клітин у полі зору) встановлено ймовірний діагноз IgG4-ЗЗ (дві ознаки із трьох згідно з діагностичними критеріями H. Umehara та співавторів (2017)) [19]. Пацієнтці було призначено лікування ГК. Але хвора уникала постійного прийому ГК, епізодично приймала преднізолон курсами по 5–7 днів у різних дозах (40; 20; 10; 5 мг), намагаючись знайти мінімально «ефективну дозу». Дійсно, періорбітальний набряк значно зменшувався на 2–3-й день, але після відміни ГК через 2 тиж знову появлявся, часто ще більший від вихідного (рис. 7).
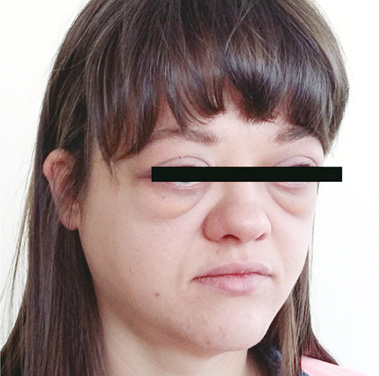
У зв’язку з персистуванням симптомів, відсутністю стійкого ефекту від епізодичного прийому ГК, а також приєднання задишки хвора звернулась для повторного обстеження. На КТ органів грудної порожнини (квітень 2016): розсіяні вогнища ущільнення легеневої тканини типу «матового скла» (ознаки фокального пневмоніту); посилення інтерстицію у прикореневих ділянках; збільшені паратрахеальні та пахвові лімфовузли. Імунологічне дослідження крові: IgG4 >300 мг/дл (при нормі 4–86 мг/дл). Загальний аналіз крові: нейтрофільний лейкоцитоз (12,2⋅10/л). КТ щелепно-лицевої ділянки з внутрішньовенним контрастуванням (жовтень 2016): приєдналося дифузне збільшення ЗМОЯ, особливо латерального прямого, без ураження їх сухожиль; прогресування збільшення слинних і слізних залоз; збереження ознак хронічного синуситу. Враховуючи прогресування захворювання, пацієнтці рекомендовано терапію ГК і метотрексатом. Але хвора відмовилася від прийому метотрексату, оскільки планувала вагітність. Тому їй було призначено ГК у дозі 40 мг/добу з повільним зниженням дози через 2–4 тиж після початку терапії (по 10 мг кожні 2 тиж до досягнення дози 20 мг, а потім зі зниженням — по 5 мг кожні 2 тиж) згідно з міжнародними консенсусними рекомендаціями щодо ведення пацієнтів із IgG4-ЗЗ [8]. Після досягнення дози преднізолону 5 мг/добу рекомендовано підтримувальну терапію протягом 1–3 років. У подальшому, залежно від можливості досягнення ремісії та зниження дози ГК, планується розглянути можливість лікування RTX.
Висновок
Цей клінічний випадок демонструє множинність проявів IgG4-ЗЗ (IgG4-пов’язані сіаладеніт, дакріоцистит, міозит орбіти, пневмоніт, лімфаденопатія) за відсутності специфічних змін з боку традиційних методів обстеження. Ключовими для встановлення діагнозу були результати імуногістохімічного дослідження та підвищення рівня IgG4 у сироватці крові. Прогресуючий перебіг захворювання підтвердив необхідність ранньої і тривалої терапії ГК з метою досягнення стійкого клінічного ефекту та уникнення незворотних пошкоджень органів.
Список використаної літератури
- 1. Сорока Н.Ф., Григорчук И.П. (2015) Иммуноглобулин G4-связанное заболевание. Здравоохранение, 7: 17–26.
- 2. Яременко О.Б., Петелицька Л.Б. (2015) IgG4-залежне захворювання — нова нозологічна одиниця в ревматології. Здоров’я України, 4(35): 56–58.
- 3. Carruthers M.N., Khosroshahi A., Augustin T. et al. (2015) The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann. Rheum. Dis., 74: 14–18.
- 4. Chen Y., Zhao J., Feng R. et al. (2016) Types of Organ Involvement in Patients with Immunoglobulin G4-related Disease. Chin. Med. J. (Engl.), 129(13): 1525–1532.
- 5. Ebbo M., Grados A., Samson M. et al. (2017) Long-term efficacy and safety of rituximab in IgG4-related disease: Data from a French nationwide study of thirty-three patients. PLoS ONE, 12(9): e0183844.
- 6. Hamano H., Kawa S., Horiuchi A. et al. (2001) High serum IgG4 concentrations in patients with sclerosing pancreatitis. N. Engl. J. Med., 344(10): 732–738.
- 7. Karim F., Loeffen J., Bramer W. et al. (2016) IgG4-related disease: a systematic review of this unrecognized disease in pediatrics. Pediatr. Rheumatol. Online J., 14: 18.
- 8. Khosroshahi A.М., Wallace Z.C., Crowe J.L. (2015) International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol., 67(7): 1688–1699.
- 9. Kleger A., Seufferlein T., Wagner M. et al. (2015) IgG4-Related Autoimmune Diseases: Polymorphous Presentation Complicates Diagnosis and Treatment. Dtsch. Arztebl. Int., 112(8): 128–135.
- 10. Lang D., Zwerina J., Pieringer H. (2016) IgG4-related disease: current challenges and future prospects. Ther. Clin. Risk Manag., 12: 189–199.
- 11. Mattoo H., Mahajan V.S., Maehara T. et al. (2016) Clonal expansion of CD4+ cytotoxic T lymphocytes in patients with IgG4-related disease. J. Allergy Clin. Immunol., 138(3): 825–838.
- 12. Moutsopoulos H.M., Fragoulius G.E., Stone J.H. (2016) Overview of IgG4-related disease. In: UpToDate, Post TW (Ed.), UpToDate, Waltham, MA.
- 13. Sebastian A., Sebastian M., Misterska‑Skóra M. et al. (2017) The variety of clinical presentations in IgG4‑related disease in Rheumatology. Rheumatol. Int., 36: 1–7.
- 14. Sedhom R., Sedhom D., Strair R. (2017) IgG4-related disease: A mini-review. J. Rare Dis. Res. Treat., 2(2): 18–23.
- 15. Stone J.H. (2018) Oral presentation at the annual meeting of the American College of Rheumatology (24 October, Chicago).
- 16. Stone J.H., Zen Y., Deshpande V. (2012) IgG4-Related Disease. N. Engl. J. Med., 366: 539–551.
- 17. Tiegs-Heiden C.A., Eckel L.J., Hunt C.H. et al. (2014) Immunoglobulin G4–Related Disease of the Orbit: Imaging Features in 27 Patients. Am J of Neuroradiol., 35 (7): 1393–1397.
- 18. Uchida K., Masamune A., Shimosegawa T., Okazaki K. (2012) Prevalence of IgG4-Related Disease in Japan Based on Nationwide Survey in 2009. Int. J. Rheum., 358371.
- 19. Umehara H., Okazaki K., Nakamura T. et al. (2017) Current approach to the diagnosis of IgG4-related disease – Combination of comprehensive diagnostic and organ-specific criteria. Mod. Rheumatol., 27(3): 381–391.
- 20. Vasilyev V., Gorodetskiy V., Safonova T. et al. (2017) Spectrum of the diseases with orbital involvement in rheumatology: single-center study. Ann. Rheum. Dis., 76: 424–425.
- 21. Vikse J., Haland S., Norheim K.B. (2017) IgG4-related disease. Tidsskr. Nor. Legeforen, 137: 1–8.
- 22. Wallace Z.S., Mattoo H., Carruthers M. et al. (2015) Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann. Rheum. Dis, 74(1): 190–195.
- 23. Weindorf S.C., Frederiksen J.K. (2017) IgG4-Related Disease: A Reminder for Practicing Pathologists. Arch. Pathol. Lab. Med., 141: 1476–1483.
- 24. Wolfson A., Hamilos D.L. (2017) Recent advances in understanding and managing IgG4-related disease. F1000Res., 6: 185.
IgG4-ЗАВИСИМОЕ ЗАБОЛЕВАНИЕ: СОВРЕМЕННОЕ СОСТОЯНИЕ ПРОБЛЕМЫ И ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ
Резюме. В статье представлен обзор данных литературы о клинических проявлениях IgG4-зависимого заболевания, особенностях диагностики и современных подходах к лечению. Описан клинический случай IgG4-зависимого заболевания у пациентки с поражением периорбитальных тканей, слезных и больших слюнных желез.
IgG4-зависимое заболевание, периорбитальный отек, слюнные железы, слезные железы, глюкокортикоиды.
Адреса для листування:
Яременко Олег Борисович
01601, Київ, бульв. Тараса Шевченка, 13
Національний медичний університет ім. О.О. Богомольця,
кафедра внутрішньої медицини № 3
Leave a comment