СУЧАСНІ МІШЕНІ ДЛЯ ЦІЛЬОВОЇ ТЕРАПІЇ РЕВМАТОЇДНОГО АРТРИТУ: ВІД МОНОКЛОНАЛЬНИХ АНТИТІЛ ДО БЛОКАТОРІВ СИГНАЛЬНИХ МОЛЕКУЛ
Коваленко В.Н., Головач И.Ю., Борткевич О.П.
Резюме. Резюме. В оглядовій статті представлено сучасні підходи до фармакотерапії ревматоїдного артриту (РА), еволюцію нових лікарських препаратів: від моноклональних антитіл до фактора некрозу пухлини-α до інгібіторів малих молекул. Розшифровка патогенезу РА, відкриття багатьох нових молекул і сигнальних шляхів, що беруть участь у патогенезі цього захворювання, уможливила створення лікарських препаратів, орієнтованих на конкретні молекули, сигнальні шляхи чи посередники активації імунної системи, що впливають на основні клінічні симптоми РА, механізми прогресування і забезпечують основну мету лікування — ремісію. Наведено характеристику нових біологічних препаратів, що впливають на В-клітини, костимуляцію Т-клітин, а також нових мішеней фармакологічного впливу — інгібіторів малих молекул. Детально описано механізм внутрішньоклітинної сигнальної трансдукції, дана характеристика молекулам сигнальних шляхів, а також представлено новий потенційно перспективний напрямок у лікуванні РА за допомогою блокаторів JAK і Syk.
Ревматоидный артрит (РА) является хроническим системным иммунозависимым воспалительным заболеванием с многофакторной этиологией [6, 24]. Патогенез, известный на сегодняшний день, представляет собой аббератную активацию клеток иммунной системы (Т- и В-лимфоциты, нейтрофилы, макрофаги, фибробласты), приводящую к последовательной активации провоспалительных цитокинов, хемокинов протеолитических ферментов, к их гиперпродукции, индуцирующих в конечном итоге костную и хрящевую деструкцию [10, 42]. Функция клеток иммунной системы, принимающих участие в развитии ревматоидного воспаления, регулируется специфическими сигнальными путями, в реализации которых участвуют множество внутриклеточных молекул [60]. Расшифровка патогенеза РА, открытие многих новых молекул и сигнальных путей, участвующих в патогенезе, возродила надежду создать лекарственные препараты, ориентированные на конкретные молекулы, сигнальные пути или посредники активации иммунной системы, способные влиять на основные клинические симптомы РА, механизмы прогрессирования и приводящие к основной цели лечении РА — ремиссии [13, 17, 28, 56].
Еще два десятилетия назад имеющиеся методы лечения были неэффективными для большинства пациентов с РА и с другими воспалительными заболеваниями суставов. Врачи регистрировали снижение качества и продолжительности жизни вследствие неконтролированной воспалительной активности РА, рост коморбидных и асоциированных состояний, оказывающий существенное влияние на прогноз, побочные эффекты применения препаратов. Отсрочка с началом лечения базисными противоревматическими препаратами через 5–7 лет оборачивалась быстрой инвалидизацией пациентов. Имеющиеся в то время болезньмодифицирующие противоревматические препараты (DMARDs) могли замедлить прогрессирование заболевания, но не могли остановить его. Эта неудовлетворительная ситуация существенно изменилась в конце 90-х годов ХХ в., когда возможности лечения для пациентов с РА были расширены за счет появления нового класса лекарственных препаратов — блокаторов фактора некроза опухоли (ФНО)-α [37, 57]. Эти препараты были призваны не только значительно снизить воспалительную активность РА, но и остановить рентгенографическую прогрессию. Этому прорыву в терапевтических возможностях способствовали новаторские исследования профессора сэра Равиндера Майни и профессора сэра Марка Фельдмана, работавших в Институте Кеннеди в Лондоне, которые представили первые химерные моноклональные антитела к ФНО-α для лечения пациентов с РА [27].
Ингибиторы ФНО-α коренным образом изменили весь терапевтический подход не только для РА, но и для анкилозирующего спондилита (АС) и псориатического артрита (ПсА). Прежде всего, они положили начало таргетной терапии, направленной на контроль прогрессирования заболевания, и убедительно доказали возможность достижения ремиссии или низкой активности у пациентов с РА [37]. Эти препараты подарили надежду сотням тысяч пациентам на выздоровление или же жесткий контроль болезни, а также воодушевили исследователей идти дальше по пути создания новых лекарственных препаратов. Именно внедрение ингибиторов ФНО-α в клиническую практику привело впоследствии к пересмотру стратегии и менеджмента больных РА [56].
В настоящее время ингибиторы ФНО-α эффективно подавляют и контролируют воспаление при этих заболеваниях, тем самым предотвращают необратимые повреждения тканей и предупреждают инвалидность пациентов [9, 37]. Рентгенологические исследования показывают, что прогрессивное повреждение суставов может быть остановлено при определенных условиях, но, к сожалению, не у всех пациентов. Использование комбинированной терапии ингибиторами ФНО-α и болезньмодифицирующими противоревматическими препаратами (DMARDs), в частности метотрексата, на ранней стадии РА способно повысить эффективность терапии, остановить или, по крайней мере, значительно замедлить прогрессирование болезни [37, 57]. Таким образом, биологическая терапия в настоящее время повсеместно одобрена для лечения РА и считается высокоэффективной, однако, эти препараты по-прежнему не в состоянии обеспечить достижение ремиссии у большинства пациентов.
Большинство имеющихся на сегодня биологических препаратов для лечения РА являются моноклональными антителами против цитокинов или хемокинов. ФНО-α — ключевой цитокин, ответственный за развитие воспаления синовиальной оболочки сустава при РА [1, 10, 27, 57]. ФНО-α, который преимущественно продуцируется активированными моноцитами и макрофагами, вызывает продукцию других противовоспалительных цитокинов, стимулирует экспрессию молекул адгезии эндотелиальными клетками и транспорт лейкоцитов в воспаленный сустав, повышает синтез металлопротеиназ, ингибирует синтез протеогликанов хряща, стимулирует активность остеокластов [19]. Таким образом, ФНО-α является медиатором развития как хронического синовита, так и деструктивного компонента ревматоидного воспаления. Сегодня в нашем арсенале есть 3 класса ингибиторов ФНО-α: моноклональные антитела (инфликсимаб, адалимумаб, голимумаб), рекомбинантная гибридная молекула рецептор/Fc-иммуноглобулин (этанерцепт) и пегилированный Fab’ фрагмент IgG1 (цертолизумаб пегол). Сегодня, по оценкам производителей, более 1 136 000 пациентов применяли и продолжают использовать инфликсимаб, 500 000 пациентов — этанерцепт, 370 000 пациентов во всем мире — адалимумаб [61]. И хотя ингибиторы ФНО-α в настоящее время считаются золотым стандартом биопрепаратов для пациентов с воспалительными артритами, есть еще ряд нерешенных вопросов о том, как получить максимальную пользу от этих препаратов [18]. Самые последние руководства ACR о том, что пациенты с ранним РА не являются кандидатами на биологическую терапию [52], остается спорным. Существуют убедительные данные, что применение ингибиторов ФНО-α на ранних стадиях болезни может быть очень эффективным и может вызвать наступление клинической ремиссии у определенной доли больных [18, 25, 64]. Семейные врачи и другие специалисты здравоохранения должны быть осведомлены о ранних симптомах воспалительных артритов, с акцентом на важность скорейшего направления таких пациентов к ревматологам для диагностики и наискорейшего начала адекватного лечения.
Хорошо также известно, что от 21 до 35% пациентов прекращают применение блокаторов ФНО-α в течение первого года [16]. Причины этого, по-видимому, связаны с отсутствием ответа, снижением эффективности, частичной эффективностью, развитием побочных эффектов, недостаточной приверженностью пациентов лечению, а в нашей стране — и с материальными трудностями. А значит, остается еще много вопросов, связанных с эффективностью и безопасностью биологических препаратов [1].
Однако РА — гетерогенное, с точки зрения патогенетических механизмов, заболевание, и гиперпродукция ФНО-α являются не единственным механизмом воспаления и тканевой деструкции при РА [7, 10]. Поэтому продолжаются исследования патогенеза РА и поиск новых мишеней для оптимального терапевтического воздействия [6, 7, 55].
Ритуксимаб. В-клетки также имеют важное значение в патогенезе РА, хотя их роль не так хорошо изучена, как Т-клеток. Однако за последние годы выяснено, что В-клетки могут участвовать в патогенезе РА не только как продуценты аутоантител (в частности ревматоидного фактора), но и как антигенпрезентирующие клетки — представляя артритогенный аутоантиген Т-клеткам. В результате Т-клетки активируются и продуцируют провоспалительные цитокины [14].
При РА в зоне воспаления суставов образуются эктопические ростковые лимфоидные структуры, состоящие из агрегатов Т- и В-лимфоцитов [62]. При этом клетки, инфильтрирующие синовиальную оболочку (синовиоциты, дендритные клетки и др.), синтезируют факторы, влияющие на выживаемость, функциональную активность и миграцию В-клеток. К ним относятся фактор, активирующий В-клетки (BAFF), лиганд, индуцирующий пролиферацию (APPRIL), CTХ лиганды хемокинов (-12 и -13) и лимфотоксин-β, интерферон (ИФН)-γ, интерлейкин (ИЛ)-5 и -15, ФНО-α. Наиболее сильной антигенпрезентирующей способностью обладают В-лимфоциты, синтезирующие ревматоидные факторы, которые могут презентовать иммунные комплексы Т-лимфоцитам, независимо от типа антигена [12]. Все это делает В-клетки перспективными терапевтическими мишенями при РА. В процессе созревания стволовых клеток в плазматические клетки В-лимфоциты проходят несколько последовательных стадий, для каждой из которых характерна экспрессия определенных дифференцировочных мембранных антигенов. CD20 — клеточный мембранный антиген, экспрессия которого характерна для ранних и зрелых В-лимфоцитов, но не стволовых, ранних преВ-, дендритных и плазматических клеток. Полагают, что истощение (деплеция) В-лимфоцитов под действием ритуксимаба реализуется за счет комбинации нескольких механизмов, включающих комплементзависимую клеточную цитотоксичность, антителозависимую клеточную цитотоксичность и индукцию апоптоза [23, 35] (рис. 1).
Таким образом, ритуксимаб является чрезвычайно эффективным и относительно безопасным препаратом для лечения РА и может рассматриваться как прообраз нового направления в лечении аутоиммунных заболеваний человека, в основе которого лежит модуляция В-клеточного звена иммунитета [12, 14].
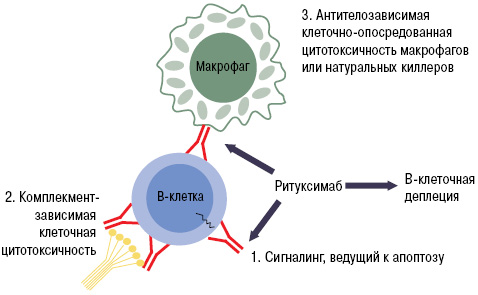
Рис. 1. Механизм действия ритуксимаба (адаптировано нами по: Cohen S.B. et al., 2006 [23])
Абатацепт. Согласно современным представлениям, Т-клетки также имеют фундаментальное значение в развитии РА, поэтому одним из важных направлений в лечении этого заболевания является подавление патологической активации Т-лимфоцитов [33, 71]. Установлено, что для оптимальной активации Т-лимфоцитов требуется, как минимум, два сигнала. Один из них реализуется в процессе взаимодействия Т-клеточных рецепторов с молекулами главного комплекса гистосовместимости (ГКГ), которые экспрессируются на мембране антигенпрезентирующих клеток (АПК), другой — за счет взаимодействия так называемых костимулирующих рецепторов на Т-клетках и соответствующих лигандов на АПК [72]. Ключевой костимуляторный сигнал обеспечивается за счет взаимодействия CD28 на Т-лимфоцитах и СD80/СD86 на АПК. CD28 постоянно экспрессируется на наивных CD4+ и CD8+ Т-клетках, а CD80 и CD86 — только после стимуляции АПК. При наличии обоих сигналов Т-лимфоциты подвергаются пролиферации и синтезируют цитокины, которые, в свою очередь, активируют другие клетки иммунной системы, и прежде всего макрофаги. В отсутствие костимуляторного сигнала Т-лимфоциты теряют способность эффективно отвечать на антигенные стимулы и подвергаются апоптозу. Наиболее мощным физиологическим ингибитором взаимодействия CD28–CD80/CD86 является CTLA4 (cytotoxic T-lymphocyte-associated antigen 4) — рецептор для CD80/CD86, который экспрессируется после активации АПК и взаимодействует с этими лигандами с более высокой авидностью (примерно в 500–2500 раз выше), чем CD28. Эта молекула рассматривается, как негативный регуляторный рецептор, который ограничивает неконтролируемую активацию Т-клеток в процессе иммунного ответа.
Эти патогенетические данные послужили основанием для разработки нового препарата абатацепт, представляющего собой растворимую гибридную белковую молекулу, состоящую из двух компонентов — внеклеточного домена CTLA4 человека и модифицированного Fc (CH2 и CH3 области) фрагмента IgG1. Модификация Fc фрагмента обеспечивает низкую способность абатацепта индуцировать комплементзависимые и антителозависимые клеточные цитотоксические реакции. Как и нативный CTLA4, этот белок связывается с более высокой авидностью с CD80/86, чем с CD28, и блокирует активацию Т-клеток (рис. 2).
По данным экспериментальных исследований, абатацепт подавляет развитие коллагенового артрита (классическая лабораторная модель РА у человека) при введении препарата во время иммунизации лабораторных животных коллагеном и замедляет прогрессирование артрита при введении на фоне его развития [68].
Абатацепт был одобрен в США и Европе в 2005 г. для лечения РА у взрослых пациентов с неадекватным ответом на DMARDs или ингибиторы ФНО-α. В январе 2010 г. он был одобрен в Европе для ювенильного артрита умеренной и тяжелой степени у больных в возрасте ≥6 лет.
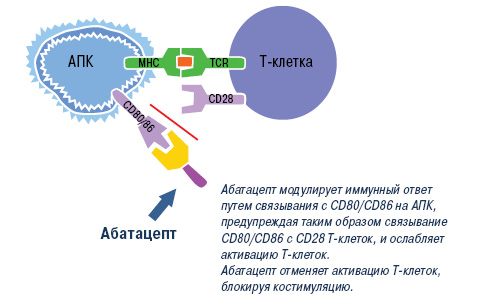
Рис. 2. Механизм действия абатацепта. TCR — рецептор Т-клетки; МНС — большой комплекс гистосовместимости
В исследовании R. Westhovens и соавторов (2009) [70] была продемонстрирована долгосрочная эффективность и безопасность абатацепта в течение 5 лет в дозе 10 мг/кг массы тела. Препарат хорошо переносился, способствовал реальному снижению активности заболевания, был безопасен. Эти данные в сочетании с относительно высоким уровнем приверженности лечению обеспечивают клинические преимущества абатацепта при РА. Кроме того, продемонстрировано, что абатацепт обеспечивал клинический эффект у пациентов с РА, ранее применявших блокаторы ФНО-α без достижения целевых результатов; при этом абатацепт был эффективен у всех пациентов, независимо от ингибитора ФНО-α, которые применяли на предыдущем этапе лечения. Этот факт позволяет предположить, что переход на абатацепт может оказаться полезным и для пациентов, не ответивших на лечение ингибиторами ФНО-α.
Тоцилизумаб.
Еще одним цитокином, обладающим плейотропным действием и играющим важную роль в патогенезе РА, является ИЛ-6. Он синтезируется многими клетками (Т- и В-лимфоцитами, моноцитами, фибробластами, эндотелиальными клетками и др.), участвующими в развитии воспаления, и проявляет широкий спектр провоспалительных биологических эффектов [2, 7, 44]. ИЛ-6 осуществляет передачу внутриклеточного сигнала двумя путями: связывание с мембранным ИЛ-6-рецептором (мИЛ-6Р) и транссигнализация (trans-signalling) [51]. При этом внутриклеточная часть мИЛ-6Р не участвует в передаче сигнала. Для этого необходим другой белок — gp130 (ИЛ-6Р β-цепь, CD130), который присутствует в клетках, не экспрессирующих мИЛ-2 грн. Наряду с мИЛ-6Р существует растворимая форма рецептора ИЛ-6 (рИЛ-6Р), которая образует комплекс с ИЛ-6, обладающий способностью связываться с gp130 и индуцировать передачу активационного сигнала (транссигнализация). В то время как классические эффекты ИЛ-6 ограничены действием на клетки, экспрессирующие мИЛ-6Р (гепатоциты, моноциты, макрофаги и некоторые субпопуляции лимфоцитов), транссигнализация позволяет ИЛ-6 активировать клетки, лишенные мИЛ-6Р, но экспрессирующие gp130, в том числе синовиальные клетки. Это лежит в основе широкого спектра патологических эффектов ИЛ-6 при РА — лихорадки, повышения концентрации острофазовых белков, анемии, синтеза аутоантител, формирования паннуса и деструкции суставов, активации Th17-клеток и др. [11].
Тоцилизумаб представляет собой гуманизированные моноклональные антитела (IgG1), которые связываясь с мембранными и растворимыми рецепторами ИЛ-6, ингибируют оба сигнальных пути ИЛ-6-зависимой клеточной активации [47] (рис. 3). Тоцилизумаб — первый и единственный препарат, обладающий способностью подавлять ИЛ-6-зависимые воспалительные реакции, разрешенный к применению при заболеваниях человека.
В настоящее время программа клинических исследований тоцилизумаба включает около 4 тыс. больных РА, что позволяет составить определенное впечатление не только об эффективности, но и о безопасности этого препарата [36].
Тоцилизумаб при применении в монотерапии или в комбинации с метотрексатом продемонстрировал превосходство монотерапии в сравнении с метотрексатом и способствовал статистически значимому снижению активности РА в течение 24 нед [36]. Тоцилизумаб также продемонстрировал эффективность у пациентов с РА, не достигших адекватного ответа или прервавших лечение ингибиторами ФНО-α [26].
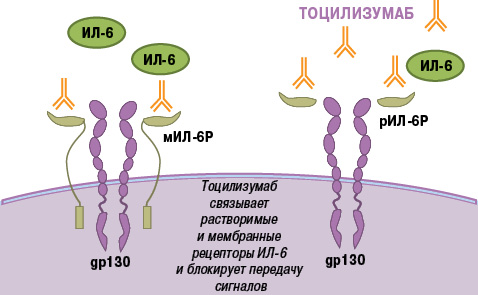
Рис. 3. Механизм действия тоцилизумаба (адаптировано нами с интернет-ресурса http://www.roactemra.com [4])
Существует тесная связь между нормализацией сывороточного уровня ИЛ-6 после лечения тоцилизумабом и клинической ремиссии. В III фазе исследования SATORI показано, что пациенты, достигшие нормализации сывороточного уровня ИЛ-6, достигли также DAS28-ремиссии. Нормализация уровня ИЛ-6 может быть адекватным маркером для выявления пациентов, которые могут остановить применение тоцилизумаба без риска активации заболевания [45].
В 3-летнем расширенном исследовании SAMURAI у пациентов с ранним РА, получавших тоцилизумаб, установлено значительное торможение рентгенографической прогрессии [61]. Более того, рентгенографическое прогрессирование эффективнее подавлялось у пациентов, применявших тоцилизумаб в начале заболевания в качестве первого биологического препарата, чем у тех, кто вначале получал DMARDs. Раннее введение тоцилизумаба в лечении может быть более эффективным в предотвращении повреждения суставов. Результаты исследования LITHE с участием 1196 пациентов, имевших недостаточный ответ на метотрексат, также подтвердили возможность тоцилизумаба подавлять рентгенографическую прогрессию [39].
Цертолизумаба пэгол. Это совершенно новый препарат группы ингибиторов ФНО-α. Формально он относится к группе человеческих моноклональных антител, однако содержит не полное моноклональное тело, а только его Fab-фрагмент (антигенсвязывающий) и не содержит комплемент-связывающий Fc-фрагмент, который присутствует в комплексном антителе. Таким образом, за счет своей уникальной структуры цертулизумаба пэгол, в отличие от инфликсимаба и адалимумаба, не связывает комплемент. Цертулизумаба пэгол также не стимулирует дегрануляцию нейтрофилов и апоптоз моноцитов или лимфоцитов периферической крови человека in vitro. Важной особенностью цертулизумаба пэгол является то, что Fab-фрагмент моноклонального тела в его составе связан с полиэтиленгликолем (ПЭГ), который существенно меняет фармакокинетику препарата. Структура препарата схематически представлена на рис. 4.
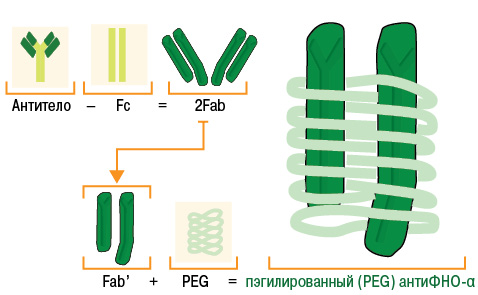
Рис. 4. Схема структуры цертолизумаба пэгола (адаптировано нами по: CIMZIA, 2009 [20])
Конъюгация Fab-фрагмента с ПЭГ в составе цертолизумаба пэгола позволила достичь повышения гидрофильности и биодоступности, а также повышения устойчивости к комплементу и нейтрализующим антителам. ПЭГ активно связывает молекулы воды, что вызывает формирование «водного облака» вокруг модифицированной молекулы «ПЭГ — белок», поэтому пэгилированные белки слабо проникают в здоровые ткани и задерживаются в кровеносном русле, накапливаются в воспаленных тканях [65].
Цертолизумаба пэгол был одобрен для лечения РА в комбинации с метотрексатом в Соединенных Штатах и Европе в 2009 г. Применение пэгилирования увеличивает период полураспада молекулы и устраняет химерные Fc-части, что обусловливает отсутствие активации комплемента [5, 65]. Поэтому есть повод надеяться, что добавление ПЭГ будет способствовать более прочному соединению с меньшим количеством побочных эффектов, хотя это еще предстоит установить, действительно ли пэгилирование предоставляет эти преимущества в реальной клинической практике [61].
Результаты исследования RAPID1 показали высокую эффективность лечения комбинацией метотрексата и цертолизумаба пэгола в обеих дозировках (200 мг и 400 мг), существенно превосходящую монотерапию метотрексатом [38]. В исследовании RAPID2 к 24-й неделе лечения 20% улучшение по критериям ACR в группах метотраксат + цертолизумаба пэгол 200 мг и метотраксат + цертолизумаба пэгол 400 мг и в группе монотерапии метотрексатом было достигнуто в 57,3; 57,6 и 8,7; 50% улучшение — в 32,5; 33,1 и 3,1; 7% улучшение — в 15,9; 10,6 и 0,8% случаев соответственно [58]. Следует обратить внимание на то, что в связи с особенностями протокола в исследование RAPID1 было включено 172 (17,5%) больных [38], а в RAPID2 — 101 (16%) больной с положительным туберкулиновым тестом (папула ≥5 мм) [38, 58].
Цертолизумаба пэгол продемонстрировал высокую эффективность при РА, сопоставимую с таковой у других ингибиторов ФНО-α, и удовлетворительную переносимость. В двух последовательных исследованиях RAPID выявлены следующие положительные моменты применения цертолизумаба пэгол: высокая стабильность ответа на лечение, быстрое развитие клинического эффекта и возможность прогнозирования ответа на терапию в течение первых 12 нед, низкая частота локальных реакций после инъекционного введения [5, 58].
Более 100 цитокинов и хемокинов вовлечены в воспалительный каскад, связанный с воспалительными артритами. Более глубокое понимание патофизиологии РА привело к выявлению новых терапевтических мишеней, в том числе провоспалительных цитокинов, Т- и В-клеток, молекул адгезии, хемокинов, внутриклеточных и внеклеточных сигнальных путей. Такие цитокины, как ИЛ-6, -12, -15, -17, -18, -21, -23, -33, ИФН-γ, являются потенциальными целями для иммуномодуляции [41], также как и сигнальные трансдукционные системы, обеспечивающие связывание цитокинов с клеточными рецепторами, а также внутриклеточные киназы, ответственные за сигнальную внутриклеточную трансмиссию.
Сегодня разрабатывается широкий спектр препаратов потенциальной терапии во многочисленных клинических испытаниях, инициированных сразу же после определенных открытий в области знаний о патогенезе РА. Эти теоретические исследования послужили мощным стимулом для разработки противовоспалительных препаратов нового поколения.
Передача сигналов от рецепторов на геном — одна из наиболее актуальных и интенсивно развивающихся областей клеточной и молекулярной биологии. Особое внимание уделяется сведениям о веществах, которые обеспечивают или блокируют частично или полностью передачу сигнала. Они представляют собой низкомолекулярные (<1 кДа) химически синтезированные вещества, получившие название малых молекул (small molecules) и предназначенные для перорального приема [13, 59]. Эти молекулы могут воздействовать на несколько рецепторов, блокируя маршруты внутриклеточных сигнальных путей, играющих важную роль в патогенезе РА [43]. Особый интерес вызывают малые молекулы, ингибирующие процессы внутриклеточной сигнализации, к ключевым компонентам которой относятся тирозин-киназы: JAK (janus kinase — янус киназа), Syk (spleen tyrosine kinase — селезеночная тирозин-киназа) и MAPK (mitogen-activatеd protein kinase — митогенактивированная протеинкиназа) [50, 66].
Передача сигналов между клетками (сигнальная трансмиссия) может быть эндокринной, паракринной и аутокринной. Сигналами для клеток крови служат гормоны, факторы роста, цитокины, хемокины и т.п. Процесс преобразования сигнала на пути от клеточной мембраны к ядру называется сигнальной трансдукцией [3].
К компонентам сигнальной трансдукции относятся молекулы, воспринимающие сигнал на поверхности клетки (мембранные рецепторы), и разнообразные молекулы-эффекторы (исполнители), которые с помощью белок-белковых взаимодействий передают этот сигнал к ядру, активируя на конечном этапе специфичные факторы транскрипции. Рецептор и связанные с ним эффекторы формируют сигнальный путь (signalling pathway). Между разными сигнальными путями за счет использования идентичных эффекторов может происходить перекрестное соединение (интеграция сигналов), в результате чего в клетке образуются так называемые сигнальные сети (signalling network). Поэтому сигнальную трансдукцию можно представить как скоординированную эстафету, в которой информация, полученная от внеклеточных сигналов, передается к внутриклеточным эффекторам, и далее — к ядру. Для успешного прохождения любого сигнала необходимо, чтобы все компоненты сигнального пути были собраны в нужном месте и именно в то время, когда это необходимо [53].
Если рассматривать систему сигнальной трансдукции в общем виде, то выявляется определенная закономерность. На входе в систему существует множество лигандов и их рецепторов (известно >200 видов рецепторов). На выходе из системы активируется несколько сотен факторов транскрипции, мишенью которых являются около 20 тыс. генов. При этом сегодня известно всего около 30 внутриклеточных сигнальных путей, которые совместно используются рецепторами разных типов [3]. Пример хорошо изученных на сегодня сигнальных путей, образующих сигнальную внутриклеточную сеть, представлен на рис. 5. Цитокины, связываясь с этими рецепторами, могут активировать различные пути сигнальной трансдукции: JAK-STAT(JAK-Signal Transducer and Activator of Transcription — сигнальный трансдуктор и активатор транскрипции), Syk, МАРК, PI3K (фосфоинозитид-3´-киназа) и др. Именно открытие нового семейства тирозин-фосфорилированных транскрипционных факторов, семейства сигнальных трансдукторов и активаторов транскрипции позволяет лучше понимать эффекты цитокинов [8, 53].
В последнее время появляются все новые факты о значимости сигнальной системы JAK-STAT в развитии целого ряда аутоиммунных заболеваний [54]. Это тем более важно с учетом того, что разработка лечебных стратегий будущего требует знания и понимания тонких механизмов патогенеза аутоиммунной патологии, в основе которой лежит нарушение клеточной сигнализации. Получены доказательства участия JAK также в патогенезе онкологических заболеваний, неврологических расстройств, диабета и сердечно-сосудистых болезней [22, 54].
JAK — небольшие молекулы (молекулярная масса 120–130 кДa), ассоциированные с цитоплазматическим участком трансмембранных рецепторов, имеют тирозин-киназный домен, ответственный за их ферментную активность [8]. Такое название — JAK — в честь двуликого бога Януса — JAK получили из-за того, что, в отличие от других протеинкиназ, они имеют два киназных домена (один из них, правда, неактивен). Основной мишенью JAK является семейство цитоплазматических факторов транскрипции STAT, которые после активации перемещаются в ядро и активируют транскрипцию своих генов-мишеней [3]. Передача внеклеточных сигналов происходит при этом на цитокиновые рецепторы (см. рис. 5). Семейство JAK в клетках млекопитающих, в противоположность остальным семействам киназ, является малочисленным и представлено только четырьмя JAK-белками: JAK1, JAK2, JAK3 и TYK2, которые были идентифицированы в начале 1990-х годов [17, 21, 40, 53]. Вскоре после их открытия была установлена их функциональная роль в передаче сигналов от интерферонов и цитокинов. По современным представлениям, именно сигнальная система JAK-STAT является ключевым компонентом регуляции иммунитета и гемопоэза. Именно через нее опосредуются эффекты многочисленных цитокинов (табл. 1). Особое значение в иммунопатологии РА придают JAK3, которая экспрессируется в первую очередь в клетках иммунной системы (В- и Т-лимфоциты, естественные киллерные клетки, моноциты). JAK3 осуществляет трансдукцию внутриклеточного сигнала от рецепторов цитокинов: ИЛ-2Р, -4Р, -7Р, -9Р, -13Р, -15Р и -21Р [28, 46, 59].
| JAK1-JAK3 | JAK1-TYK2 | JAK1-TYK2-JAK2 | JAK1-JAK2 | JAK2-TYK2 | JAK2-JAK2 | ||
|---|---|---|---|---|---|---|---|
| ИЛ-2, -4, -17, -15, -21 |
ИФН | ИЛ-10, -20, -22 |
ИЛ-6, -11, -27; G-CSF |
ИФН-γ | ИЛ-12, -23 | EPO, TRO, GH, PRL | ИЛ-3, -5 |
| • Рост и созревание лимфоидных клеток • Дифференциация и поддержание гомеостаза Т-клеток, натуральных киллеров • Переключение В-клеток • Воспаление |
• Антивирусный • Воспалительный • Антиопухлевой |
• Дифференциация Т-клеток • Т-клеточный гомеостаз • Воспаление • Гранулопоэз |
• Антивирусный •Воспалительный |
• Врожденный иммунитет •Дифференциация и пролиферация Th17-клеток • Воспаление |
• Эритропоэз • Миелопоэз • Продукция тромбоцитов • Рост • Развитие молочных желез |
||
G-SCF — гранулоцитарный колониестимулирующий фактор, EPO — эритропоэтин, TRO — трофинин, GH — гормон роста, PRL — пролактин, TYK — тирозин киназа.
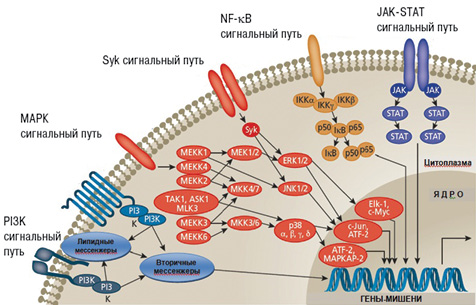
Рис. 5. Схема внутриклеточных сигнальных каскадов, играющий патогенетическую роль при РА (адаптировано нами по: Mavers M. et al., 2009 [40]). Воздействие цитокинов, хемокинов, ростовых факторов, патогенассоциированных молекулярных паттернов, эндогенных факторов (стресс, ультрафиолетовое облучение) приводит к активации рецепторов на поверхности клеток, что запускает внутриклеточный сигнальный каскад. Внутриклеточный сигналинг, в свою очередь, обусловливает изменение экспрессии генов, участвующих в воспалении, деградации внеклеточного матрикса, апоптозе и других важных клеточных процессах и устанавливает соответствующее реагирование на раздражители. ATF — активированный транскрипционных фактор; Elk — эукариотподобная протеинкиназа; ERK — экстрацеллюлярная сигналзависимая киназа; IKK — ингибитор каппа В киназы; JNK — c-Jun N-терминальная киназа; STAT — сигнальный трансдуктор и активатор транскрипции; NF-kB — нуклеарный (ядерный) фактор каппа В
Таким образом, именно JAK3 может служить полезной мишенью при РА [22, 34]. Важным обоснованием для применения ингибиторов JAK3 при РА являются данные о более выраженной экспрессии JAK3, STAT1, STAT4 и STAT6 в синовиальной ткани пациентов с РА, чем у лиц с остеоартрозом и спондилоартритом [67]. Имеются данные о том, что in vitro ингибитор JAK3 подавляет синтез ФНО-α, ИФН-γ, ИЛ-6 и -8, матриксной металлопротеиназы (ММП)-3 и пролиферацию активированных CD4+ Т-лимфоцитов, ИЛ-17 Th17-клетками и ИФН-γ Th1-клетками [31, 63].
Кроме того, на разных стадиях проводимые исследования по изучению эффективности и безопасности нескольких ингибиторов малых молекул (табл. 2).
| Агент | Мишень внутриклеточного сигнального пути |
Фаза исследования |
|---|---|---|
| Тофаситиниб (Tofacitinib, ТОФА, СР-690,550) | JAK 1,2,3 | Май 2012, разрешен FDA к применению |
| Фостанатиниб (Fostanatinib, R788) | Syk | 3 |
| LY3009104 (INCB28050) | JAK1,2 | 2/3 |
| VX-509 | JAK3 | 2 |
| GLPG-0259 | MAPK-APK5 | 2 |
| GLPG-0634 | JAK1 | 2 |
В мае 2012 г. FDA разрешил к клиническому применению новый препарат тофаситиниб (Tofacitinib; CP-690,550; «Pfizer»), который ингибирует (IC50) JAK3 [74]. В настоящее время в программу клинических исследований тофаcитиниба включено более 5000 пациентов (около 5700 пациенто-лет) [29]. Завершены 4 исследования Ib–II фазы, в которых подбиралась эффективная и безопасная доза препарата. Программа III фазы рандомизированных плацебо-контролируемых клинических исследований (РПКИ) включает 6 исследований. В настоящее время опубликованы данные 5 РПКИ, в которых сравнивали эффективность монотерапии тофаcитинибом, комбинированной терапии тофаcитинибом и метотрексатом у пациентов, резистентных к метотрексату, базисным противовоспалительным препаратам (DMARDs) и ингибиторам ФНО-α. Лечение тофаcитинибом в дозе ≥3 мг 2 раза в сутки привело к быстрому результату со значительной эффективностью по сравнению с плацебо; при оценке первичной конечной точки (ACR20 ответ на 12-й неделе) ответ достигнут в 39,2% (3 мг; p≤0,05), 59,2% (5 мг; р<0,0001), 70,5% (10 мг; р<0,0001) и 71,9% (15 мг; р<0,0001) в группе тофаcитиниба и 35,9% пациентов в группе адалимумаба (р=0,105) по сравнению с 22,0% пациентов, получавших плацебо. Улучшения зарегистрированы на 24-й неделе, в соответствии с ACR20-, ACR50- и ACR70-ответами, а также классификации ремиссии. Наиболее распространенными нежелательными реакциями у пациентов во всех группах лечения тофацитинибом (n=272) были инфекции мочевыводящих путей (7,7%), диарея (4,8%), головная боль (4,8%) и бронхит (4,8%). Таким образом, монотерапия тофацитинибом в дозе ≥3 мг 2 раза в сутки была эффективна при лечении пациентов с активным РА в течение 24 нед и продемонстрировала приемлемый профиль безопасности [29].
Еще одной потенциальной мишенью «терапии будущего» является Syk [32, 55]. Она экспрессируется преимущественно в В-клетках и в меньшей степени — в тучных клетках, макрофагах, нейтрофилах и синовиальных фибробластах. Syk вовлечена в процесс внутриклеточной сигнализации посредством многокомпонентных иммунных рецепторов, включающих Iga (В-клеточный рецептор), g-цепь Т-клеточного рецептора, FcgР, интегрины, которые содержат так называемый иммунный рецептор тирозиновый активационный мотив (immune-receptor tyrosine-based activation motif — ITAM). Активация этих рецепторов приводит к фосфорилированию ITAM, что, в свою очередь, индуцирует фосфорилирование Syk, приводящее к активации многочисленных путей сигнализации, регулирующих процессы пролиферации и синтеза цитокинов [13, 40, 49]. Поскольку Fc и рецепторы В-клеток являются важными в патогенезe аллергических и иммунных заболеваний, а также выявлено значительное повышение уровня фосфорилированной Syk в синовиальной ткани при РА в сравнении с пациентами с остеоартрозом, блокирование Syk сигнального пути может быть эффективной терапевтической мишенью [43]. Ингибитор Syk, скорее всего, ослабит эффекторную фазу, блокируя В-клеточную рецепторную сигнализацию и FcgR-сигнализацию через нейтрофилы, присутствующие в синовиальной оболочке. Кроме того, ингибирование Syk имеет дополнительное преимущество в предотвращении созревания остеокластов, ослаблении узурации, разрушении суставов и остеопении, ассоциированной с РА [43, 59].
Фостаматиниб (FosD, Fostamatinib; «Rigel»/«AstraZeneca») является пролекарством; после перорального приема он быстро превращается в R406, который является мощным селективным ингибитором Syk. Данные экспериментальных исследований свидетельствуют о том, что фостаматиниб эффективно подавляет развитие синовита, эрозий и паннуса при коллагеновом артрите у мышей [48]. При этом подавление экспрессии Syk коррелирует со снижением синтеза некоторых цитокинов (ИЛ-1 и -6), лиганда хемокинов KC-GRO-α, металлопротеиназ и макрофагального хемоаттрактантного белка 1 [32].
В настоящее время эффективность и безопасность фостаматиниба изучаются в рамках 3 РПКИ III фазы: OSKIRA-1 (недостаточная эффективность метотрексата), OSKIRA-2 (недостаточная эффективность DMARDs), OSKIRA-3 (лечение метотрексатом и недостаточная эффективность одного из ингибиторов ФНО-α).
В двойном слепом плацебо-контролируемом исследовании установлено эффективность и безопасность фостаматиниба у больных с активным РА, несмотря на терапию метотрексатом [69]. На 12-й неделе терапии был достигнут ответ по ACR20 65 и 72% при приеме 100 и 150 мг фостаматиниба соответственно, в сравнении с плацебо 38% (p<0,01). Наиболее частыми побочными эффектами были диарея и нейтропения.
MAPК киназы также являются ключевыми регуляторами активности и продукции цитокинов. MAPK фосфорилируют сериновые, треониновые и тирозиновые аминокислотные остатки внутриклеточных сигнальных белков, регулирующих выживаемость и пролиферацию клеток, синтез цитокинов и металлопротеиназ. Описано три основных субсемейства MAPK, каждое из которых регулирует несколько каскадных реакций (см. рис. 5). К ним относятся JNK, ERK и p38 [13, 66, 73]. Все они экспрессируются синовиальной тканью, а, значит, могут быть потенциальными мишенями терапевтического воздействия. Наибольшее значение при РА имеет путь, опосредованный р38. MAPK p38 представляет собой наиболее «дистальный» сигнальный этап, предшествующий активации факторов транскрипции, которые регулируют гены основных провоспалительных цитокинов (ФНО-α; ИЛ-1, -6, -8), а также циклооксигеназы (ЦОГ)-2 и коллагеназы-1 и -3 [17]. Кроме того, в синовиальной ткани р38 регулируют синтез металлопротеиназ, участвующих в деструкции сустава, остеокластогенез, функции эндотелия, созревание дендритных клеток, экспрессию костимуляторных молекул Т-лимфоцитов. Все это вместе взятое позволило предположить, что ингибиция р38 может быть эффективным подходом к подавлению воспаления при РА [73].
Однако практически все ингибиторы р38 (на сегодня их известно >22) [17, 28, 30], по данным исследований I/II фазы, оказались неэффективными и токсичными у пациентов с РА. Наиболее подробно изучен селективный ингибитор изоформы р38 MAPK памапимод (Pamapimod; RO4402257; «Hoffmann-La Roche»). Однако этот препарат оказался менее эффективным, чем метотрексат, а частота побочных эффектов была такая же, как у метотрексата [15].
Кроме ингибиторов сигнальных молекул, активно исследуются антагонисты цитокинов и хемокинов, например ингибиторы ИЛ-12/ИЛ-23 (Апилимод месилат; Apilimod mesylate; STA-5326), анти-CCR5 (Маравирок; Maraviroc), анти-CXCL-10 (MDX-1100), ингибиторы транскрипционных факторов [43, 55, 59, 66]. Проводятся исследования II фазы препаратов Апремиласт (Apremilast, CC-10004 — ингибитор фосфодиэстеразы), CF101 (агонист А3-рецепторов аденозина). Отрицательные результаты получены при изучении ARRY-438162 (ингибитор MEK1 и MEK2), CE-224, 535 (селективный антагонист P2X7) и ERB-041 (селективный агонист эстрогеновых рецепторов). Займут ли эти новые препараты ведущее место в лечении РА и смогут ли кардинально изменить подходы к лечению этого заболевания, — покажет время.
Разработка ингибиторов сигнальных молекул является ярким примером высочайших достижений медицины и фармакологии в ревматологии в XXI в. Потенциальными достоинствами этих препаратов могут быть удобный для пациентов пероральный прием и более низкая стоимость, чем моноклональных антител. Однако для окончательного вывода о месте ингибиторов сигнальных молекул в лечении РА необходимы дальнейшие исследования. Они, в первую очередь, должны быть направлены на изучение их безопасности в процессе длительного применения в реальной клинической практике, у пациентов с различными формами РА и коморбидными заболеваниями [13].
Подводя итог, необходимо отметить, что РА является тяжелым недугом. Сегодня мы гораздо больше знаем о патофизиологии, иммунных нарушениях при РА, чем несколько десятилетий назад. Понимая это, мы смогли определить новые мишени терапии, в том числе влияние на цитокины, хемокины, клетки иммунной системы и киназы, ингибирование которых оказалось весьма эффективным в целевом лечении РА. С помощью этих методов терапии мы смогли достичь очень хороших результатов в различных аспектах заболевания: регрессирование признаков и симптомов, сохранение структурной целостности, возобновление функциональной способности, а самое главное — возможность достижения ремиссии у значительного бо`льшего числа больных [40]. Появилась возможность стратифицировать пациента по тяжести заболевания и персонифицировать терапию РА. Имея в арсенале новые препараты и используя их в своей практике, мы можем оптимизировать результаты лечения больных РА [17, 21, 43, 59, 66]. И в настоящее время перед ревматологами стоит задача, как наилучшим образом интегрировать передовые лечебные технологии в повседневную клиническую практику.
СПИСОК использованной ЛИТЕРАТУРЫ
1. Балабанова Р. М. (2010) Инфликсимаб: на все ли вопросы получены ответы за 10 лет? Совр. ревматол.,1: 61–65.
2. Головач И.Ю. (2011) Ревматоидный артрит: достижения биологической терапии и интерпретация клинических исследований. Рационал. фармакотер., 4: 29–34.
3. Домнинский Д.А. (2011) Молекулярные механизмы лейкозогенеза. Механизмы реализации сигнальной трансдукции. Онкогематология, 1: 76-84.
4. Интернет-ресурс: http://www.roactemra.com/portal/roactemra/mechanism_of_action
5. Каратеев Д.Е., Насонов Е.Л., Денисов Л.Н. и др. (2012) Новые возможности терапии ревматоидного артрита: российский опыт применения цертолизумаба пэгола. Науч.-практ. ревматол., 51(2): 14–19.
6. Коваленко В.М. (2009) Ревматичні захворювання: сучасні тенденції фармакотерапії. Укр. ревматол. журн., 3(37): 5–11.
7. Коваленко В.Н., Головач И.Ю., Борткевич О.П. (2011) Индивидуализация лечения ревматоидного артрита: курс на достижение оптимальных результатов. Укр. ревматол. журн., 3(45): 5–15.
8. Минеев В.Н., Сорокина Л.Н. (2006) Современные представления о JAK-STAT системе как новой сигнальной системе и ее нарушениях при иммунной патологии: механизмы негативной регуляции. Ч. II. Аллергология, 1: 47–55.
9. Насонов Е.Л. (2003) Моноклональные антитела к фактору некроза опухоли в ревматологии. РМЖ, 11: 390–394.
10. Насонов Е.Л. (2005) Фармакотерапия ревматоидного артрита — взгляд в 21 век. Клин. медицина, 6: 8–12.
11. Насонов Е.Л. (2009) Новые возможности фармакотерапии ревматических болезней — ингибирование интерлейкина 6. Клин. фармакол. тер., 1: 60–67.
12. Насонов Е.Л. (2006) Перспективы применения моноклональных антител к В-лимфоцитам (ритуксимаб) при ревматоидном артрите. Клин. фармакол. терапия, 15: 55–58.
13. Насонов Е.Л., Денисов Л.Н., Станислав М.Л. (2012) Новые аспекты фармакотерапии ревматоидного артрита: ингибиторы малых молекул. Науч.-практ. ревматол., 2: 66–75.
14. Проценко Г.А. (2009) Перспективы применения ритуксимаба в ревматологии. Укр. ревматол. журн., 1(35): 44–47.
15. Alten R.E., Zerbini C., Jeka S. et al. (2010) Efficacy and safety of pamapimod in patients with active rheumatoid arthritis receiving stable methotrexate therapy. Ann. Rheum. Dis., 69: 364–367.
16. Blom M., Kievit W., Fransen J. et al. (2009) The reason for discontinuation of the first tumor necrosis factor (TNF) blocking agent does not influence the effect of a second TNF blocking agent in patients with rheumatoid arthritis. J. Rheumatol., 36: 2171–2177.
17. Bonilla-Hernán M.G., Miranda-Carús M.E., Martin-Mola E. (2011) New drugs beyond biologics in rheumatoid arthritis: the kinase inhibitors. Rheumatology (Oxford)., 50(9): 1542–1550.
18. Breedveld F.C., Weisman M.H., Kavanaugh A.F. et al. (2006) The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum., 54: 26–37.
19. Choy E.H., Panayi G.S. (2001) Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med.,344: 907–916.
20. CIMZIA (summary of product characteristics) (2009). Bruxelles, UCB Pharma, Belgium, SA.
21. Cohen S. (2012) Small molecular therapies for rheumatoid arthritis: where do we stand? Expert Opin. Invest. Drugs, 21: 23–31.
22. Cohen S., Fleischmann R. (2010) Kinase inhibitors: a new approach to rheumatoid arthritis. Curr. Opin. Rheumatol., 22: 330–336.
23. Cohen S.B., Emery P., Greenwald M.W. et al. (2006) Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum., 54: 2793–2806.
24. Cutolo M. (2007) Developments in Treatments for Rheumatoid Arthritis. Eur. Musculoskelet. Rev., 12: 22–24.
25. Emery P., Breedveld F.C., Hall S. et al. (2008) Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet, 372: 375–382.
26. Emery P., Keystone E., Tony H. et al. (2008) IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial (RADIATE study). Ann. Rheum. Dis., 67: 1516–1523.
27. Feldmann M., Maini R.N. (2002) Discovery of TNF-α as a therapeutic target in rheumatoid arthritis: preclinical and clinical studies. Joint Bone Spine,69:12–18.
28. Fleischmann R. (2012) Novel small-molecular therapeutics for rheumatoid arthritis. Curr. Opin. Rheumatol., 24(3): 335–341.
29. Fleischmann R., Cutolo M., Genovese M.C. et al. (2012) Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum., 64(3): 617–629.
30. Genovese M.C. (2009) Inhibition of p38: has the fat lady sung? Arthritis Rheum., 60: 317–320.
31. Ghoreshi K., Jesson M., Li X. et al. (2011) Modulation oh innate and adaptive immunity responses by tofacitinib. J. Immunol., 186: 423442–423443.
32. Gómez-Puerta J.A., Bosch X. (2011) Spleen tyrosine kinase inhibitors — novel therapies for RA. Nat. Rev. Rheumatol., 7(3): 134–136.
33. Isaacs J.D. (2008) Therapeutic T-cell manipulation in rheumatoid arthritis: past, present and future. Rheumatology, 47: 1461–1468.
34. Ivanenkov Y.A., Balakin K.V., Lavrovsky Y. (2011) Small molecule inhibitors of NF-kB and JAK/STAT signal transduction pathways as promising anti-inflammatory therapeutics. Mini Rev. Med. Chem., 11(1): 55–78.
35. Johnson P., Glennie M. (2003) The mechanism of action of rituximab in the elimination of tumor cells. Semin. Oncol., 30 (2): 38.
36. Jones G. (2010) The AMBITION trial: tocilizumab monotherapy for rheumatoid arthritis. Expert Rev. Clin. Immunol., 6:189–195.
37. Kalden J.R. (2011) Anti-TNF therapy: what have we learned in 12 years? Arthritis Res.Ther., 13 (Suppl. 1): S1.
38. Keystone E., van der Heijde D., Mason D. et al. (2008) Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: findings of a fifty-two-week, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum.,58: 3319–3329.
39. Kremer J.L., Blanco R., Brzosko M. et al.(2011) Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate at 1 year: the LITHE study. Arthritis Rheum., 63: 609–621.
40. Mavers M., Ruderman E.M., Perlman H. (2009) Intracellular signal pathways: potential for therapies. Curr Rheumatol. Rep., 11(5): 378–385.
41. McInnes I.B., Schett G. (2012) The pathogenesis of rheumatoid arthritis. New Engl. J., 365: 2205–2219.
42. Mclnnes I.B., Schett G. (2007) Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol., 7: 429–442.
43. Navarro-Compán V., Navarro-Sarabia F. (2011) Safety and Efficacy of the Newer Biological Therapeutics in the Treatment of Rheumatoid Arthritis. Clin. Med. Insights: Therap., 3: 195–203.
44. Nishimoto N., Kishimoto T. (2006) Interleukin 6, from bench to bedside. Nat Clin Praс. Rheumatol.,11: 619–626.
45. Nishimoto N., Miyasaka N., Yamamoto K. et al. (2009) Study of active controlled tocilizumab monotherapy for rheumatoid arthritis patients with an inadequate response to methotrexate (SATORI): significant reduction in disease activity and serum vascular endothelial growth factor by IL-6 receptor inhibition therapy. Mod. Rheumatol.,19:12–19.
46. Okamoto H., Kobayashi A. (2011) Tyrosine kinases in rheumatoid arthritis. J. Inflam., 8: 21.
47. Oldfield V., Dhillon S., Plosker G.L. (2009) Tocilizumab. A review of its use in the management of rheumatoid arthritis. Drugs, 69: 609–632.
48. Pine P.R., Chang B., Schoettler N. et al. (2007) Inflammation and bone erosion are suppressed in models of rheumatoid arthritis following treatment with a novel SyK inhibitor. Clin. Immunol., 124: 244–257.
49. Riccaboni M., Bianchi I., Petrillo P. (2010) Spleen tyrosine kinases: biology, therapeutic targets and drugs. Drug Discovery Today, 15(13–14): 517–530.
50. Riese R.J., Krishnaswami S., Kremer J. (2010) Inhibition of JAK kinases in patients with rheumatoid arthritis: scientific rationale and clinical outcomes. Best Pract. Res. Clin. Rheumatol., 24: 513–526.
51. Rose-John S., Scheller J., Elson G. et al. (2006) Interleukin-6 is coordinated by membranebound and soluble receptors: role in inflammation and cancer. J. Leukocyte Biol., 80: 227.
52. Saag K.G., Teng G.G., Patkar N.M. et al. (2008) American College of Rheumatology: American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum., 59: 762–784.
53. Scott J., Pawson T. (2009) Cell signaling in space and time: where proteins come together and when they’re apart. Science, 326: 1220–1224.
54. Seavey M.M., Dobrzanski P. (2012) The many faces of Janus kinase. Biochem. Pharmacol., 83(9): 1136–1145.
55. Senott L., Vencovsky J., Pavelka K. et al. (2009) Prospective new biological therapies for rheumatoid arthritis. Autoimmun. Rev., 9: 102–107.
56. Smolen J., Aletaha D., Bijlsma J.W.J. et al. (2010) Treating rheumatoid arthritis to target: recommendations of an international task force. Ann. Rheum. Dis., 69: 631–637.
57. Smolen J.S., Emery P. (2011) Infliximab: 12 years of experience. Arthritis Res.Ther., 13(Suppl. 1): S1.
58. Smolen J.S., Landewe R., Mease P. et al.(2009) Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann. Rheum. Dis., 68: 797–804.
59. Stanczyk J., Ospelt C., Gay S. (2008) Is there a future for small molecule drugs in the treatment of rheumatic diseases? Curr. Opin.Rheumatol., 20(3): 257–262.
60. Sweeney S.E., Firestein G.S. (2007) Primer: signal transduction in rheumatic disease — a clinician’s guide. Nat. Clin. Pract. Rheumatol., 3: 651–660.
61. Tak P., Kalden J.R. (2011) Advances in rheumatology: new targeted therapeutics. Arthritis Res.Ther., 13(Suppl. 1): S5.
62. Takemura S., Klimiuk P.A., Braun A. et al. (2001) T cell activation in rheumatoid synovium is B cell dependent. J. Immunol.,167: 4710–4718.
63. Tanaka Y., Yamaoka K. (2011) Cytokine-mediated signaling as targets for treatment of rheumatoid arthritis: a JAK kinase inhibitor in vitro and in vivo. Int. J. Clin. Rheumatol., 6: 439–444.
64. van Vollenhoven R.F., Ernestam S., Geborek P. et al. (2009) Addition of infliximab compared with addition of sulfasalazine and hydroxychloroquine to methotrexate in patients with early rheumatoid arthritis (SWEFOT trial): 1-year results of a randomized trial. Lancet, 374: 459–466.
65. Voulgari P.V. (2008) Emerging drugs for rheumatoid arthritis. Expert Opin. Emerging Drugs., 13: 175–196.
66. Waldburger J.M., Firestein G.S. (2009) Garden of therapeutic delights: new targets in rheumatic diseases. Arthritis Res. Ther., 11: 206–212.
67. Walker J.G., Ahern M.J., Coleman M. et al. (2006) Expression of Jak3, STAT1, STAT4, and STAT6 in inflammatory arthritis: unique Jak3 and STAT4 expression in dendritic cells in seropositive rheumatoid arthritis. Ann. Rheum. Dis., 65: 149-156.
68. Webb L.M., Walmsley M.J., Feldmann M. (1996) Prevention and amelioration of collagen–induced arthritis by blockade of the CD28 co-stimulatory pathway: requirement for both B7–1 and B7–2. Eur. J. Immunol., 26: 2320–2328.
69. Weinblatt M., Kavanaugh A., Burgos-Vargas R. et al. (2008) Treatment of rheumatoid arthritis with a Syk kinase inhibitor. Arthritis Rheum., 11: 3309–3318.
70. Westhovens R., Kremer J., Moreland L. et al. (2009) Safety and efficacy of the selective costimulation modulator abatacept in patients with rheumatoid arthritis receiving background methotrexate: a 5-year extended phase MB study. J. Rheumatol., 36: 736–742.
71. Weyand C., Goronzy J.J. (2006) T-cell-tаrgeted therapies in rheumatoid arthritis. Nat. Clin. Pract. Rheumatol., 2: 201–210.
72. Yamada A., Salama A.D., Sayegh M.H. (2002) The role of novel T-cell co-stimulation pathways in autoimmunity and transplantation. J. Am. Soc. Nephrol., 13: 559–575.
73. Yazici Y., Regens A.L. (2011) Promising new treatments for rheumatoid arthritis — the kinase inhibitors. Bull. NYU Hosp. Jt Dis., 69(3): 233–237.
74. Zerbini C.A., Lomonte A.B. (2012) Tofacitinib for the treatment of rheumatoid arthritis. Expert. Rev. Clin. Immunol., 8(4): 319–331.
Адрес для переписки:
Коваленко Владимир Николаевич
03680, Киев, ул. Народного ополчения, 5
ГУ «ННЦ «Институт кардиологии им. акад. Н.Д. Стражеско» НАМН Украины»
Leave a comment